
Corea&di&Hun,ngton&OMIM143100users.unimi.it/cusbio/scaricare/SanoMalatoapprof.pdf · 2016-09-27 ·...
18
Corea di Hun,ngton OMIM 143100 La mala&a di Hun,ngton (HD: Hun,ngton Disease), o Corea di Hun,ngton, è una mala&a gene,ca neurodegenera,va del sistema nervoso centrale che colpisce la coordinazione muscolare e porta ad un declino cogni,vo e a problemi psichiatrici. Le zone del cervello più colpite sono quelle correlate ai movimen, e quelle legate alla memoria a breve termine. La prima descrizione completa della mala&a fu faDa dal medico George Hun,ngton, da cui il nome, nel 1872 (Fig. 1). Frequenza La prevalenza nella popolazione Caucasica è s,mata in 1/10.0001/20.000. Esordisce ,picamente durante la mezza età; in alcuni casi i sintomi si manifestano prima dei 20 anni, con disturbi del comportamento e difficoltà di apprendimento (mala&a di Hun,ngton giovanile, JHD). E’ la mala&a gene,ca che, nei quadri clinici neurologici, si manifesta più frequentemente con movimen, involontari anomali. Ques, movimen, ricordano una danza da cui deriva il nome della mala&a: in greco choreia, significa danza. È molto più comune nelle persone di discendenza europea occidentale rispeDo a chi è di origine asia,ca o africana, questo perché, nella popolazione di discendenza europea, l’allele del gene dell’hun,ng,na con ripe,zioni di CAG tra le 28 e le 35 è molto più frequente. Uno dei più al, valori di incidenza della mala&a, si registra nelle isolate popolazioni della regione del lago di Maracaibo in Venezuela, dove la mala&a di Hun,ngton colpisce fino a 7.000 individui su 1.000.000. Altre aree ad alta prevalenza sono state trovate in Tasmania e in specifiche regioni di Scozia, Galles e Svezia. L'aumento della prevalenza, in alcuni casi, si Fig.1 Ar,colo originale pubblicato nel 1872 dal doD. George Hun,ngton.
Transcript of Corea&di&Hun,ngton&OMIM143100users.unimi.it/cusbio/scaricare/SanoMalatoapprof.pdf · 2016-09-27 ·...

Corea di Hun,ngton OMIM 143100
La mala&a di Hun,ngton (HD: Hun,ngton Disease), o Corea di Hun,ngton, è una mala&a gene,ca neurodegenera,va del sistema nervoso centrale che colpisce la coordinazione muscolare e porta ad un declino cogni,vo e a problemi psichiatrici. Le zone del cervello più colpite sono quelle correlate ai movimen, e quelle legate alla memoria a breve termine. La prima descrizione completa della mala&a fu faDa dal medico George Hun,ngton, da cui il nome, nel 1872 (Fig. 1).
Frequenza La prevalenza nella popolazione Caucasica è s,mata in 1/10.000-‐1/20.000. Esordisce ,picamente durante la mezza età; in alcuni casi i sintomi si manifestano prima dei 20 anni, con disturbi del comportamento e difficoltà di apprendimento (mala&a di Hun,ngton giovanile, JHD). E’ la mala&a gene,ca che, nei quadri clinici neurologici, si manifesta più frequentemente con movimen, involontari anomali. Ques, movimen, ricordano una danza da cui deriva il nome della mala&a: in greco choreia, significa danza. È molto più comune nelle persone di discendenza europea occidentale rispeDo a chi è di origine asia,ca o africana, questo perché, nella popolazione di discendenza europea, l’allele del gene dell’hun,ng,na con ripe,zioni di CAG tra le 28 e le 35 è molto più frequente. Uno dei più al, valori di incidenza della mala&a, si registra nelle isolate popolazioni della regione del lago di Maracaibo in Venezuela, dove la mala&a di Hun,ngton colpisce fino a 7.000 individui su 1.000.000. Altre aree ad alta prevalenza sono state trovate in Tasmania e in specifiche regioni di Scozia, Galles e Svezia. L'aumento della prevalenza, in alcuni casi, si
Fig.1 Ar,colo originale pubblicato nel 1872 dal doD. George Hun,ngton.

verifica a causa di un locale effeDo del fondatore, dovuto ad una storia di migrazione in zone di isolamento geografico. (Fig.2)
Cause La mala&a è causata da una mutazione in una delle due copie (alleli) di un gene, presente sul cromosoma 4, che codifica per la proteina chiamata hun,ng,na. Si traDa di una mutazione autosomica dominante. La base gene,ca della mala&a è stata scoperta nel 1993 grazie ad una ricerca internazionale guidata dalla Hereditary Disease Founda,on. E’ una mala&a deDa TRED, ovvero una mala&a da espansione di tripleDe (Trinucleo,de Repeat Disorders), caraDerizzate da un aumento eccessivo di ripe,zioni della tripleDa nucleo,dica CAG, che codifica per una glutammina (abbreviata in gln o Q). L’individuo è sano se ha meno di 35 ripe,zioni, è malato se ne ha più di 40. Tra 35 e 40 ripe,zioni l’individuo può sviluppare la mala&a, nel caso avvenga un’espansione del numero di tripleDe (Fig. 3). L’espansione avviene in vari stadi dello sviluppo, in diversi ,pi di cellule, ed è sensibile al sesso del genitore che la trasmeDe. Si è osservato, ad esempio, che l’allele HD trasmesso per via paterna è più propenso ad espandere il numero di tripleDe. Per questo mo,vo, un individuo con padre affeDo da HD ha più probabilità di sviluppare la mala&a di uno con madre affeDa da HD.
Fig. 2 Frequenza di HD nel mondo
Fig. 3. Tabella con le informazioni riguardanti la malattia HD e la sua localizzazione nel genoma umano.

Quando l’hun,ng,na è mutata Il gene che produce la proteina chiamata hun,ng,na (H@), è IT15 (Interes,ng Transcript 15). Viene espresso in tuDe le cellule di mammifero con concentrazioni maggiori nel cervello (nucleo striato) e nel tes,colo, e moderate quan,tà nel fegato, cuore e polmoni. L’hun,ng,na è essenziale per un regolare sviluppo embrionale, funziona come agente an,apopto,co, controlla la formazione del tubo neuronale e il faDore neurotrofico cerebrale (BDNF Brain-‐derived neurotrophic factor) che protegge i neuroni durante la neurogenesi. Le ripe,zioni eccessive di Glu nell’hun,ng,na mutata (mH@) tendono ad essere tagliate, con rilascio del frammento N-‐terminale della proteina. Ques, frammen, formano aggrega, di hun,ng,na nel cervello, diventano tossici, e intasano il proteasoma, l’organello adibito a spazzino cellulare che distrugge le proteine anomale, precedentemente marcate dall’ubiqui,na.
Gli aggrega, sono tossici per le cellule in quanto interferiscono con la trascrizione a livello nucleare e, nel citoplasma, con la trasmissione sinap,ca, il trasporto assonale e la funzione mitocondriale.
Studi recen, hanno mostrato che aggrega, di hun,ng,na mutata possono sequestrare piccole proteine dalla loro naturale localizzazione, inibendo la loro regolare funzione nelle cellule nervose. Una di queste è la CREB-‐binding protein o CBP (Fig. 4A), una proteina regolatrice della trascrizione che ha un ruolo essenziale per la cellula. Gli aggrega, di hun,ng,na rimuovono CBP dalla sua posizione sul DNA. Una volta sequestrata, la CBP non può compiere la propria funzione, ossia coadiuvare i faDori di trascrizione che a&vano geni necessari per la vita della cellula. Questo porta alla morte del neurone. L’hun,ng,na inoltre favorisce nei neuroni il trasporto delle vescicole e la trasmissione sinap,ca, mentre gli aggrega, interrompono il normale fluire delle vescicole lungo il citoscheletro con diminuito rilascio di neurotrasme&tori (Fig. 4.B). Infine, la disfunzione dei mitocondri meDe a rischio la sopravvivenza dei neuroni. I mitocondri producono ATP, la fonte di energia per la cellula e, nel cervello, mantengono l’omeostasi del calcio. Gli aggrega, di mHD agiscono sulle membrane dei mitocondri, diminuendo la soglia di calcio necessario per l‘apertura dei pori MPT (mitochondrial permeability transi,on), così avviene il rilascio del Cyt c (Citocromo C) che induce apoptosi (Fig. 4.C). E’ anche stato osservato che mHD provoca la ridoDa espressione di BDNF, il faDore neurotrofico cerebrale, contribuendo alla progressiva perdita di neuroni nel nucleo striato del cervello. Sebbene la sequenza degli amminoacidi determini la struDura tridimensionale della proteina sono necessari faDori proteici che agevolino il “folding” ossia il ripiegamento correDo della proteina. Il con,nuo monitoraggio del correDo ripiegamento viene effeDuato dalle proteine di accompagnamento deDe “chaperonine”. I prodo& mal ripiega,, fruDo del “misfolding”, sono anch’essi elimina, dal sistema proteosoma-‐ubiqui,na (vedi sopra). La mancata eliminazione delle proteine genera l’accumulo di fibrille amiloidi (struDure che sono la causa di patologie che prendono il nome di amiloidosi, Fig 5); l’amiloide è una
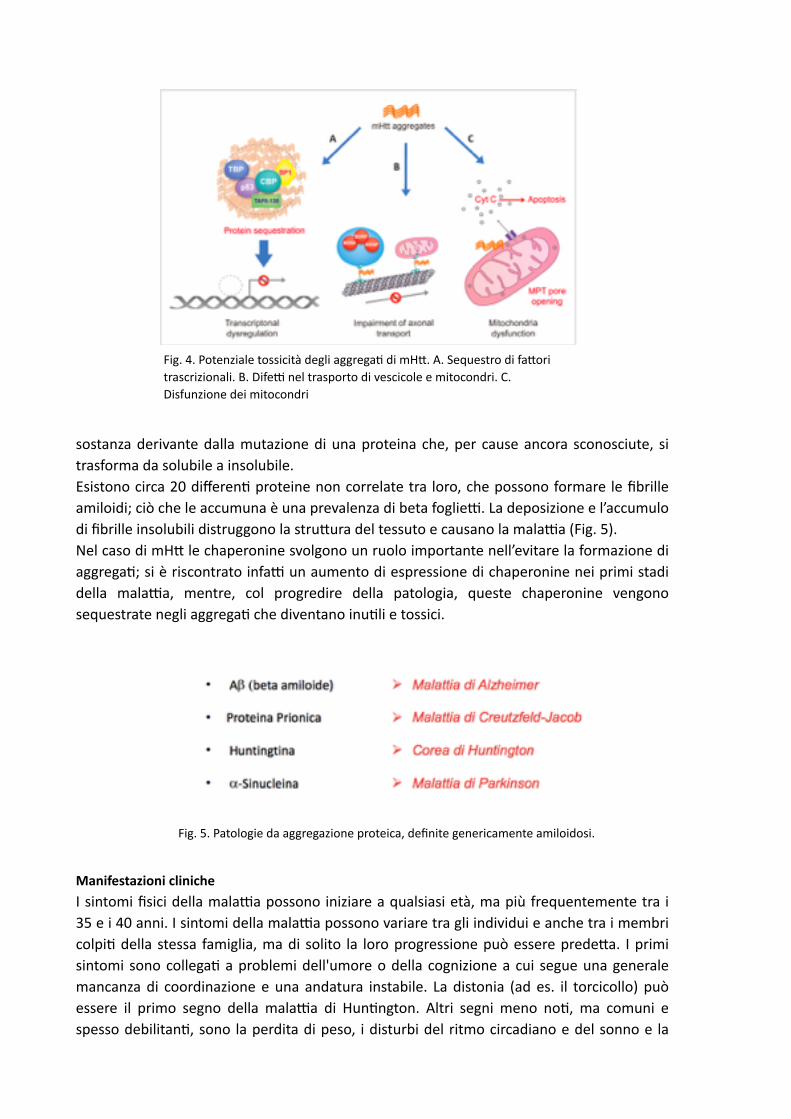
sostanza derivante dalla mutazione di una proteina che, per cause ancora sconosciute, si trasforma da solubile a insolubile. Esistono circa 20 differen, proteine non correlate tra loro, che possono formare le fibrille amiloidi; ciò che le accumuna è una prevalenza di beta foglie&. La deposizione e l’accumulo di fibrille insolubili distruggono la struDura del tessuto e causano la mala&a (Fig. 5). Nel caso di mHD le chaperonine svolgono un ruolo importante nell’evitare la formazione di aggrega,; si è riscontrato infa& un aumento di espressione di chaperonine nei primi stadi della mala&a, mentre, col progredire della patologia, queste chaperonine vengono sequestrate negli aggrega, che diventano inu,li e tossici.
Manifestazioni cliniche I sintomi fisici della mala&a possono iniziare a qualsiasi età, ma più frequentemente tra i 35 e i 40 anni. I sintomi della mala&a possono variare tra gli individui e anche tra i membri colpi, della stessa famiglia, ma di solito la loro progressione può essere predeDa. I primi sintomi sono collega, a problemi dell'umore o della cognizione a cui segue una generale mancanza di coordinazione e una andatura instabile. La distonia (ad es. il torcicollo) può essere il primo segno della mala&a di Hun,ngton. Altri segni meno no,, ma comuni e spesso debilitan,, sono la perdita di peso, i disturbi del ritmo circadiano e del sonno e la
Fig. 4. Potenziale tossicità degli aggrega, di mHD. A. Sequestro di faDori trascrizionali. B. Dife& nel trasporto di vescicole e mitocondri. C. Disfunzione dei mitocondri
Fig. 5. Patologie da aggregazione proteica, definite genericamente amiloidosi.

disfunzione del sistema nervoso autonomo. La disartria (disturbo motorio del linguaggio) e la disfagia (difficoltà a deglu,re) si accentuano durante la progressione della mala&a. Il linguaggio e la deglu,zione diventano gradualmente più problema,ci, con il rischio di soffocamento. Tu& i pazien, sviluppano ipocinesia (rallentamento o riduzione dei movimen, spontanei) e rigidità, che sfociano nella difficoltà dell'individuo ad iniziare un nuovo movimento e nella diminuzione notevole dei movimen,. Con l'avanzare della mala&a i movimen, non coordina, del corpo diventano sempre più eviden, e sono accompagna, da un calo delle capacità mentali e dalla insorgenza di problemi comportamentali e psichiatrici. Le complicanze, come la polmonite, le mala&e cardiache e i danni fisici da cadute, riducono l'aspeDa,va di vita a circa 20 anni a par,re dall'esordio dei sintomi. Fino ad oggi non esiste una cura per questa mala&a e, nelle fasi più avanzate, diventa necessaria un’ assistenza a tempo pieno. I traDamen, sono solo farmacologici e non possono alleviare mol, dei numerosi sintomi. Evoluzione Storia del gene dell’ hun,ng,na Questo gene si affaccia alla storia dell’evoluzione 800 milioni di anni fa in una par,colare ameba (Regno: pro,s, classe: protozoi): il Dictyostelium discoideum che rappresenta il primo organismo pluricellulare comparso sulla terra (ha la proprietà di passare dalla forma unicellulare a quella pluricellulare quando le condizioni sono avverse: solo in questo modo acquista mobilità) non presenta CAG nel gene dell’hun,ng,na. Il gene nasce “innocente”, ossia senza CAG (Fig. 6) nei protostomi (Celentera,, Platelmin,, Nematodi, Anellidi, Molluschi, Inse&). Nei deuterostomi (Echinodermi, Corda,: cefalocorda,, urocorda,, vertebra,) compare CAG: per esempio il riccio di mare ha due CAG.
Fig.6 Storia evolu,va della tripleDa CAG dal Dictyostelium che ne è privo, fino all’uomo
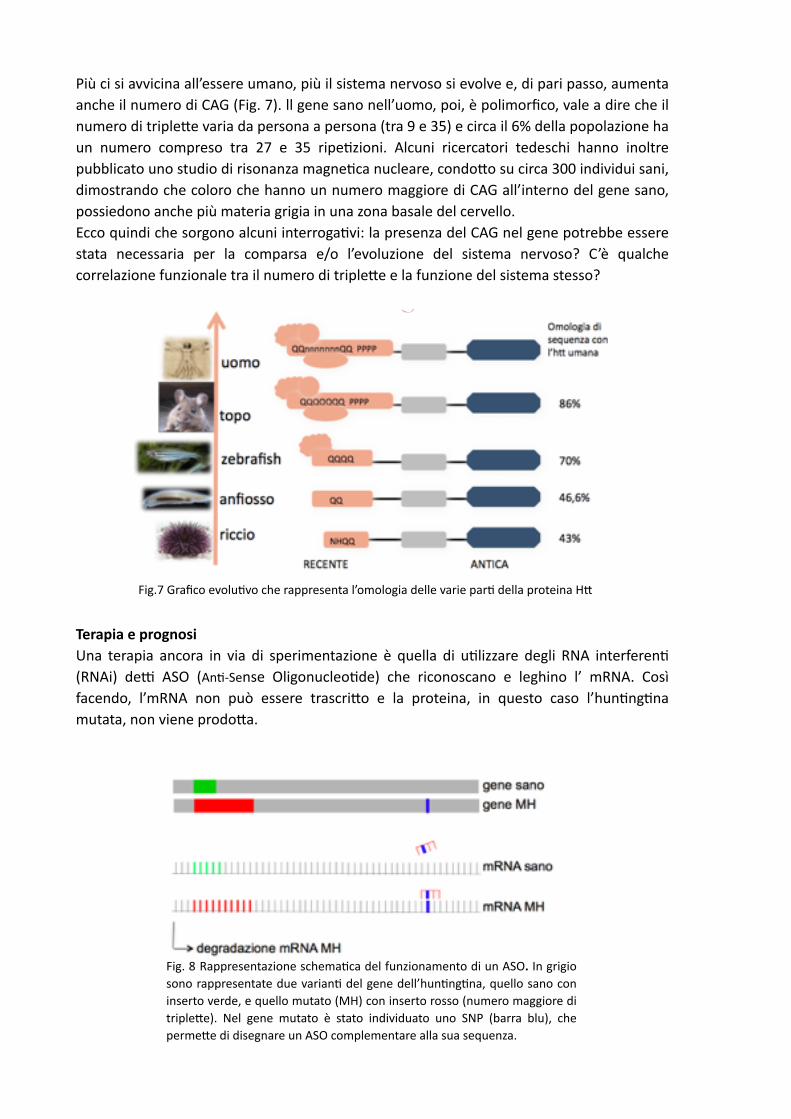
Più ci si avvicina all’essere umano, più il sistema nervoso si evolve e, di pari passo, aumenta anche il numero di CAG (Fig. 7). ll gene sano nell’uomo, poi, è polimorfico, vale a dire che il numero di tripleDe varia da persona a persona (tra 9 e 35) e circa il 6% della popolazione ha un numero compreso tra 27 e 35 ripe,zioni. Alcuni ricercatori tedeschi hanno inoltre pubblicato uno studio di risonanza magne,ca nucleare, condoDo su circa 300 individui sani, dimostrando che coloro che hanno un numero maggiore di CAG all’interno del gene sano, possiedono anche più materia grigia in una zona basale del cervello. Ecco quindi che sorgono alcuni interroga,vi: la presenza del CAG nel gene potrebbe essere stata necessaria per la comparsa e/o l’evoluzione del sistema nervoso? C’è qualche correlazione funzionale tra il numero di tripleDe e la funzione del sistema stesso?
Terapia e prognosi Una terapia ancora in via di sperimentazione è quella di u,lizzare degli RNA interferen, (RNAi) de& ASO (An,-‐Sense Oligonucleo,de) che riconoscano e leghino l’ mRNA. Così facendo, l’mRNA non può essere trascriDo e la proteina, in questo caso l’hun,ng,na mutata, non viene prodoDa.
Fig.7 Grafico evolu,vo che rappresenta l’omologia delle varie par, della proteina HD
Fig. 8 Rappresentazione schema,ca del funzionamento di un ASO. In grigio sono rappresentate due varian, del gene dell’hun,ng,na, quello sano con inserto verde, e quello mutato (MH) con inserto rosso (numero maggiore di tripleDe). Nel gene mutato è stato individuato uno SNP (barra blu), che permeDe di disegnare un ASO complementare alla sua sequenza.

Questo meccanismo di silenziamento genico potrebbe essere una buona strategia per curare mala&e monogeniche, ossia dovute alla mutazione su un solo gene. Le molecole interferen,, però, sono poco sele&ve e potrebbero riconoscere anche l’mRNA prodoDo dal gene sano. Per questo mo,vo si sta lavorando su varian, geniche. Ossia vengono cercate delle sequenze deDe SNPs (Single Nucleo,de Polymorphism), situate in un punto preciso del gene mutato. Ques, SNPs vengono u,lizza, per dis,nguere l’mRNA mutato da quello sano, aggirando il problema appena discusso. Gli SNPs vengono riconosciu, da un piccolo RNA (Fig. 8 ), che agisce con lo stesso principio del RNAi, bloccando solo l’mRNA mutato.
Atassia di Friedreich OMIM 229300
L’atassia è una mala&a neurodegenera,va che interessa il midollo spinale e il cervelleDo. Consiste nella progressiva perdita della coordinazione muscolare che quindi rende difficoltosa l'esecuzione di alcuni movimen, volontari. Esistono forme di atassia di cui non si conosce la causa (sporadiche) e forme ereditarie. Fra queste ul,me ve ne sono di autosomiche, dominan, e recessive, ma anche forme legate al cromosoma X. L’atassia (FRDA) descriDa nel 1863 dal Dr. Friedreich (Fig. 1), in famiglie con al, livelli di consanguineità, è la più frequente fra le forme autosomiche recessive. FrequenzaLa frequenza della patologia nella popolazione caucasica è s,mata in 1:29000-‐50000. L'incidenza è molto più bassa negli asia,ci e nei sogge& di discendenza africana. La frequenza dei portatori è stata s,mata essere 1:60-‐120.
Cause Nel 97% dei casi la mala&a è dovuta all’espansione della tripleDa GAA. Nel primo introne del gene FXN sul cromosoma 9 (9q21.11) gli individui sani presentano un numero di ripe,zioni da 5 fino a circa 33 volte (la maggior parte meno di 12) della tripleDa GAA. Sono portatori di premutazione i sogge& con da 34 a 65 ripe,zioni, mentre nelle persone malate, la tripleDa si trova ripetuta da 66 fino a 1700 volte (la maggior parte fra 600 e 1200). Il gene FXN codifica una proteina della famiglia delle frataxine. Si traDa di una proteina mitocondriale che lega il Ferro e ne controlla l'ingresso nei mitocondri ed è coinvolta nella respirazione cellulare. Le ripe,zioni eccessive provocano una diminuzione di livello sia degli mRNA che della proteina, che viene prodoDa solo in minima parte. La produzione di frataxina è inversamente proporzionale alle dimensioni dell’espansione. L’espansione della tripleDa GAA nel primo introne del gene FRDA influenza l’espressione stessa del
Fig. 1 Dr. Nicolaus Friedreich, neurologo e patologo tedesco che nel 186D descrisse per la prima volta la sindrome dell’atassia

gene in quanto all’ampliamento del numero di tripleDe nei sogge& mala, è associato lo stato di me,lazione delle regioni CpG (aree del DNA ricche di CG, spesso localizzate nel gene nel punto di inizio della trascrizione e che, se me,late, silenziano il gene) nell’introne 1 e questo fenomeno porta alla ridoDa trascrizione; ciò non avviene nei portatori di premutazione. Cause della mala&a sembrano essere quindi un aumento della concentrazione di ferro libero all’interno dei mitocondri che interferisce nega,vamente con l’a&vità di alcuni enzimi mitocondriali con conseguente deficit della funzione respiratoria della cellula (Fig.2). Si osserva quindi un aumento della sensibilità allo stress ossida,vo e infine la morte cellulare dovuta ai radicali liberi. Le cellule colpite sono i neuroni del midollo spinale, della corteccia e del cervelleDo.Manifestazioni cliniche I primi sintomi sono la difficoltà nella corsa e nelle a&vità spor,ve in genere, una goffaggine generale e scarsa coordinazione (atassia) durante la deambulazione. La mala&a comporta una progressiva perdita della coordinazione motoria: vengono colpi, generalmente per primi gli ar, inferiori, provocando instabilità nel cammino. Successivamente compaiono disturbi nella coordinazione delle mani e nell’ar,colazione della parola. Anche se i disturbi sono progressivi, il decorso della mala&a è variabile. Mediamente 15 anni dopo l'esordio della mala&a si ha perdita della deambulazione e mol, sono i pazien, costre& all’uso della sedia a rotelle. L'intelligenza non appare compromessa ma possono essere presen, problemi di udito, di vista (fissazione instabile dello sguardo) e di deglu,zione. Altre manifestazioni patologiche sono muscolari o scheletriche (la pianta del piede molto arcuata, la scoliosi, Fig. 3), a stadi avanza, il 20% dei pazien, sviluppa il diabete mellito. Frequen, sono i disturbi cardiaci che comportano l’aumento della frequenza cardiaca, l’ispessimento de l l e pa re, de l seDo vent r i co la re e l e a l te raz ion i dell’eleDrocardiogramma: la cardiomiopa,a è la causa più comune di morte. Il numero di ripe,zioni della tripleDa è inversamente proporzionale all'età di esordio della patologia e con il tempo che intercorre tra l'esordio e la dipendenza dalla sedia a rotelle mentre è direDamente proporzionale alla prevalenza della cardiomiopa,a.
Fig. 2 Nella matrice mitocondriale si accumula Ferro isolato da molecole di Frataxina; questo comporta un deficit della funzione respiratoria che conduce alla morte cellulare.
Fig. 3 Manifestazioni patologiche del l ’Atassia a carico del la colonna vertebrale.

Terapia e prognosiPer ridurre la sintomatologia dovuta allo stress ossida,vo vengono u,lizza, farmaci an,ossidan, che sono meno tossici e meglio tollera, rispeDo a quelli che facilitano la rimozione dell’eccesso di ferro dai mitocondri. I pazien, con cardiopa,a vengono traDa, con an,coagulan,, an,aritmici e pacema,er, mentre quelli con diabete mellito di solito necessitano dell'insulina. Assume par,colare importanza il traDamento terapeu,co riabilita,vo con l’u,lizzo di protesi e sedia a rotelle che aiutano a mantenere uno s,le di vita a&vo e può essere necessaria la logopedia. Recen, ricerche stanno sperimentando nuovi approcci terapeu,ci, basa, sul ripris,no della sintesi della frataxina. La vita media è di circa 40 anni, a seconda dell'età di esordio e della comparsa del diabete e della cardiomiopa,a. Il decesso è essenzialmente dovuto alla cardiopa,a e alla broncopolmonite. Diagnosi La conferma diagnos,ca viene effeDuata mediante l’analisi molecolare che iden,fica le mutazioni nel gene FXN. L’analisi molecolare è effeDuata sui pazien, e loro familiari oppure come test prenatale, aDraverso la villocentesi o l’amniocentesi. Si u,lizzano due metodiche che sono fra loro complementari: la PCR, rapida ma può rivelarsi problema,ca per gli individui eterozigo,, e il southern blot che permeDe di diagnos,care con precisione le mutazioni. Il southern blot prevede che il DNA, tagliato con enzimi di restrizione, venga soDoposto ad eleDroforesi su gel di agarosio e successivamente trasferito su un filtro di nitrocellulosa o nylon: l’uso di sonde radioa&ve o fluorescen, consente di iden,ficare una sequenza bersaglio perchè complementare a quella delle sonde.
Negli ul,mi anni è stata messa a punto la TP PCR (Triplet Repeat Primed PCR) che usa due coppie di primers per effeDuare l’amplificazione: i tradizionali primer forward e reverse fluorescen, e fiancheggian, la zona da amplificare e altri due primer che presentano le ripe,zioni di tripleDe (GCA)5 o (TGC)5 all’estremità 3’. Il risultato dell’amplificazione permeDe di calcolare quante ripe,zioni sono presen, e se il gene si trova nella condizione di premutazione.
Oltre all'esame del DNA, sono sta, esegui, vari ,pi di test per la valutazione della coordinazione muscolare. Ad esempio, è stato misurato il tempo impiegato dai sogge& per percorrere a piedi una determinata distanza, oppure è stato valutato quanto spesso i partecipan, riuscivano a ripetere una certa sequenza di sillabe nell'arco di dieci secondi. Un altro esperimento consisteva nell'inserire piccoli perni nei fori di una tavola e poi nell’ estrarli nuovamente, cercando di svolgere il compito il più velocemente possibile. Questo ,po di prove sono state progeDate per essere facilmente realizzate ovunque, senza il bisogno di par,colari aDrezzature tecnologiche. In aggiunta, in alcuni centri di ricerca coinvol, nello studio è stato possibile u,lizzare la risonanza magne,ca per valutare con precisione il volume totale del cervello e la dimensione delle sue singole par,.
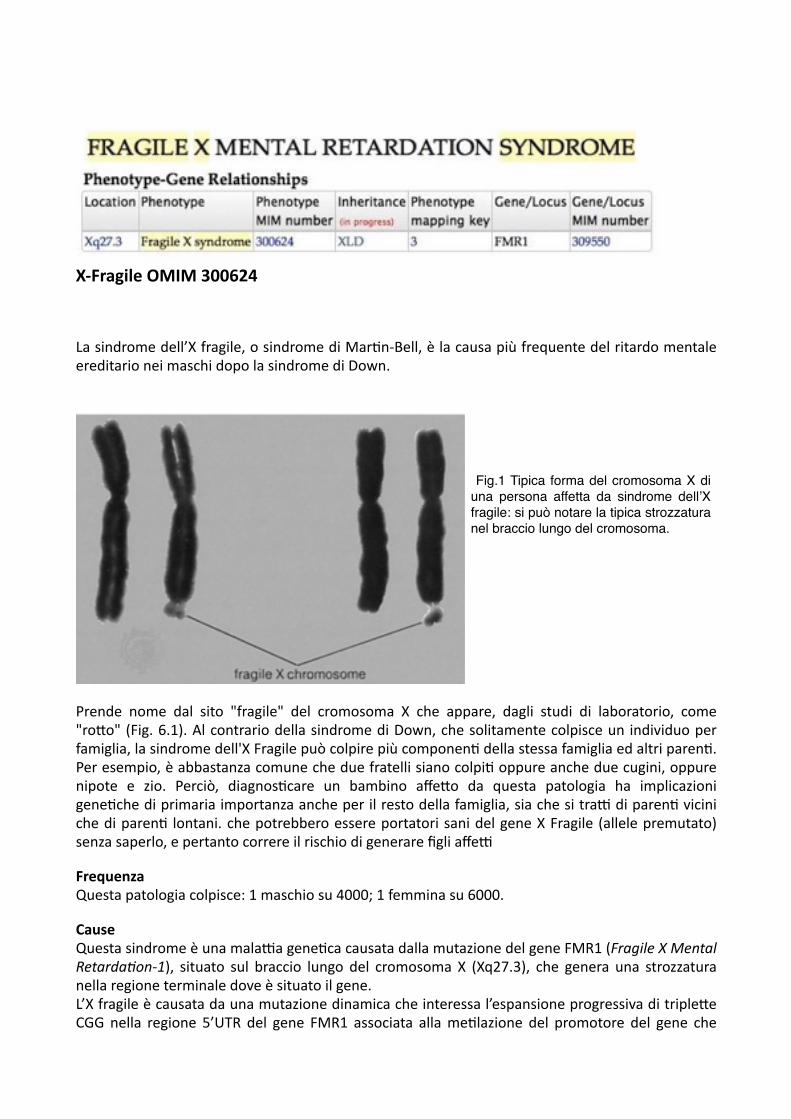
X-‐Fragile OMIM 300624
La sindrome dell’X fragile, o sindrome di Mar,n-‐Bell, è la causa più frequente del ritardo mentale ereditario nei maschi dopo la sindrome di Down.
Prende nome dal sito "fragile" del cromosoma X che appare, dagli studi di laboratorio, come "roDo" (Fig. 6.1). Al contrario della sindrome di Down, che solitamente colpisce un individuo per famiglia, la sindrome dell'X Fragile può colpire più componen, della stessa famiglia ed altri paren,. Per esempio, è abbastanza comune che due fratelli siano colpi, oppure anche due cugini, oppure nipote e zio. Perciò, diagnos,care un bambino affeDo da questa patologia ha implicazioni gene,che di primaria importanza anche per il resto della famiglia, sia che si tra& di paren, vicini che di paren, lontani. che potrebbero essere portatori sani del gene X Fragile (allele premutato) senza saperlo, e pertanto correre il rischio di generare figli affe&
FrequenzaQuesta patologia colpisce: 1 maschio su 4000; 1 femmina su 6000.
Cause Questa sindrome è una mala&a gene,ca causata dalla mutazione del gene FMR1 (Fragile X Mental Retarda7on-‐1), situato sul braccio lungo del cromosoma X (Xq27.3), che genera una strozzatura nella regione terminale dove è situato il gene. L’X fragile è causata da una mutazione dinamica che interessa l’espansione progressiva di tripleDe CGG nella regione 5’UTR del gene FMR1 associata alla me,lazione del promotore del gene che
Fig.1 Tipica forma del cromosoma X di una persona affetta da sindrome dell’X fragile: si può notare la tipica strozzatura nel braccio lungo del cromosoma.

determina il silenziamento del gene e l’assenza della proteina FMRP (Fragile X Mental Retarda7on Protein). Questa proteina, espressa sopraDuDo nei tessu, del tes,colo e del cervello, è implicata nello sviluppo delle connessioni neuronali del cervello. Il suo ruolo nel tes,colo non è stato ancora chiarito. FMRP è una proteina che lega gli RNA (RNA binding protein), si associa a mRNA codifican, importan, proteine neuronali, regolandone aspe& essenziali come il trasporto lungo i dendri, verso le sinapsi e la traduzione in proteine; in assenza di FMRP, mol, degli mRNA bersaglio sono deregola,, e maggiormente trado& in proteine. Normalmente il numero di tripleDe CGG contenute nel 5’UTR è compreso tra le 6 e le 54 copie; un’espansione tra 55 e 200 copie definisce un soggeDo premutato portatore sano di X-‐fragile, in grado di produrre la proteina; gli affe& hanno ripe,zioni della tripleDa CGG superiori a 200 copie (Fig. 2).
La trasmissione può avvenire in maniera asintoma,ca oppure può innescare un’ulteriore espansione fino a raggiungere la mutazione completa; la sindrome ha una trasmissione dominante legata al cromosoma X con penetranza ridoDa nelle femmine. Può essere trasmessa dalla madre ai propri figli (maschi o femmine) e dal padre a tuDe le figlie femmine (il gene FMR1 non può essere trasmesso dal padre ai figli maschi a cui passano solo il cromosoma Y). Il gene FMR1 può esistere nello stadio di premutazione in una famiglia per parecchie generazioni senza che causi alcuni problemi di sviluppo ma può espandersi nel passaggio da una generazione all’altra (Fig. 3). La tendenza all’espansione si verifica maggiormente quando la premutazione è trasmessa per via materna, mentre quando la premutazione è trasmessa dal padre rimane stabile.
Fig.2 localizzazione e numero di ripe,zioni della tripleDa CGG presente nella persona normale, premutata e malata.

Manifestazioni cliniche
Si accompagna comunemente a disturbi del comportamento e ad altri problemi dello sviluppo, come disfunzioni specifiche dell'apprendimento, au,smo e difficoltà comportamentali significa,ve. I sintomi della mala&a sono spesso più lievi nelle femmine rispeDo ai maschi.
La patologia non compare alla nascita, si manifesta progressivamente, dai primi mesi di vita alla pubertà. I sogge& affe& da X fragile presentano un volto allungato, padiglioni auricolari a impianto basso, ampi e anteversi. La mascella generalmente è sporgente (progna,smo), sono presen, palato ogivale, mal occlusione dentale e in alcuni sogge& strabismo oculare. Può essere presente macrocefalia (assoluta o rela,va), ipotonia, iperlassità ar,colare, cifoscoliosi, pia&smo plantare, prolasso della valvola mitrale. Il macroorchidismo (aumento di volume dei tes,coli) si presenta generalmente in fase post-‐puberale. Le bambine colpite da questa sindrome possono avere un aspeDo normale oppure presentare alcuni segni caraDeris,ci della sindrome, fra cui il viso allungato, le orecchie prominen, ed il palato alto.
L’en,tà della disabilità intelle&va è variabile, spesso i sogge& affe& da questa sindrome soffrono di ipera&vità e deficit di aDenzione. L’ipera&vità tende a diminuire con l’età, mentre le difficoltà di aDenzione restano fonte di problemi sia nell’adolescenza che nell’età adulta. Inoltre, i maschi possono mostrare inusuali aDeggiamen,, quali, ad esempio, morsicarsi le mani e le braccia o sbaDere le mani quando sono in condizioni di eccitamento o sovra s,molazione.
Spesso gli individui di sesso maschile, manifestano comportamen, di ,po au,s,co, quali ad esempio uno scarso contaDo oculare con l’interlocutore, avversione nell’essere tocca,, aDeggiamen, stereo,pa, e ripe,,vi, linguaggio ripe,,vo, rigidità negli interessi, difficoltà ad acceDare i cambiamen, della loro rou,ne abituale, faDo che provoca in loro aDacchi d’ira o problemi di comportamento e instabilità emo,va. TuDavia, solo una minoranza mostra au,smo. La maggior parte dei maschi colpi, è affeDuosa ed ha interesse a stringere relazioni sociali anche se
Fig. 3. A sinistra: trasmissione per via materna. La tendenza all’espansione si verifica maggiormente quando la premutazione è trasmessa per via materna; durante la maturazione dell’ovulo la premutazione può espandersi e diventare mutazione completa. A destra: trasmissione per via paterna. Quando la premutazione è trasmessa dal padre rimane stabile; le figlie femmine riceveranno la premutazione senza che avvengano variazioni nel numero delle tripleDe CGG. I figli maschi ricevono dal padre il cromosoma Y, pertanto non sono a rischio di ereditare la premutazione.

mostra notevoli difficoltà nelle interazioni e tende ad essere schiva ed ansiosa. I ragazzi X Fragile hanno scarse caraDeris,che di motricità fine o di controllo delle proprie mani e muscoli delle dita. Scrivere è estremamente difficile per loro, probabilmente a causa dello scarso tono muscolare, dell’iperestensibilità delle giunture delle dita e della loro limitata abilità nel pianificare e portare a termine azioni motorie fini complesse.
Le femmine con X Fragile spesso presentano meno problemi dei maschi e per questo parecchie di esse non sono mai state diagnos,cate. Alcune ragazze hanno buone capacità accademiche e possono mostrare solo leggeri problemi sociali e di comportamento, come un'eccessiva ,midezza, mentre altre possono mostrare un certo range di problemi di apprendimento e di comportamento. Solo il 30% presenta un grave ritardo mentale nell’età adulta.
Terapia e prognosi
Non esiste una terapia risolu,va. È però possibile il traDamento dei sintomi per migliorare la qualità della vita dei pazien,, combinando l’impiego di varie categorie di psicofarmaci per traDare autolesionismo, comportamento aggressivo e au,smo. Per disturbi d'ansia e ossessivo-‐compulsivi vengono usa, gli s,molan, e inibitori sele&vi della ricaptazione della serotonina (SSRI).
Data la plas,cità del cervello dei bambini, l’intervento precoce è molto importante al fine di favorire l’integrazione e il recupero. Oltre ai farmaci, devono quindi essere presi in considerazione gli interven, possibili in mol, seDori, come educazione, istruzione, riabilitazione, interven, logopedici, fisioterapici, e occupazionali, tu& dire& a migliorare l’autonomia dei pazien,.
Sono in fase di studio, come nuovi farmaci, molecole con funzione sia di antagonis, di neurotrasmeHtori eccitatori come il glutammato (uno in par,colare a&vo sul receDore mGluR5, iden,ficato dai ricercatori della Università CaDolica di Roma, è aDualmente oggeDo di sperimentazione clinica di Fase II) che di agonis, di neurotrasmeHtori inibitori (come il GABA-‐A e GABA-‐B).
I primi risulta, sono promeDen, e potrebbero modificare il decorso e migliorare la prognosi.
Recentemente è stato effeDuato uno studio usando topi modifica, gene,camente privi della proteina FMRP e che riproducevano parzialmente la sintomatologia della sindrome dell’X fragile negli umani; questa ricerca ha mostrato come il blocco dei receDori CB1 dei cannabinoidi con il farmaco Rimonabant normalizza le alterazioni cogni,ve, la sensibilità al dolore e le crisi epileHche. Questa scoperta suggerisce che la somministrazione di farmaci che bloccano l'a&vità del sistema cerebrale endocannabinoide (modulatore chiave della plas,cità sinap,ca, delle prestazioni cogni,ve, dell'ansia, della nocicezione e della susceHbilità alle crisi epile&che) potrebbe rappresentare una nuova strategia per traDare pazien, con la sindrome dell’X fragile.
Diagnosi Essendo un feno,po lieve alla nascita, la diagnosi clinica della sindrome dell’X fragile nei bambini può essere difficile. Le famiglie all’interno delle quali sia presente un soggeDo affeDo dalla sindrome possono avvalersi di una consulenza gene,ca che possa informarle sul rischio di incidenza della mala&a.

Sebbene sempre più individui e famiglie colpi, da sindrome dell'X-‐fragile vengano iden,fica,, essa rimane ancora largamente sconosciuta e soDovalutata. Eppure una diagnosi precoce è di grande importanza per le famiglie, ai fini di avere una consulenza gene,ca appropriata e quindi un intervento sullo sviluppo, che migliora le possibilità di condurre una vita produHva e soddisfacente. La diagnosi (anche prenatale sui villi coriali o sul liquido amnio,co) può essere effeDuata mediante l’analisi del cario,po che evidenzia la forma ,pica che il cromosoma X ha negli affe&; l’indagine dell’espansione della tripleDa può essere eseguita tramite test basa, sulla reazione a catena della polimerasi (PCR) e confermata mediante il metodo Southern blot (che valuta anche la me,lazione del promotore, Fig. 4). La PCR potrebbe non evidenziare la presenza di espansioni di tripleDe di grosse dimensioni: in pazien, di sesso femminile si potrebbe erroneamente diagnos,care un geno,po omozigote perchè la PCR potrebbe non aver amplificato un allele, così come per lo stesso mo,vo potrebbero non essere osservate premutazioni o mutazioni complete in pazien, di sesso maschile.
Distrofia Muscolare di Duchenne OMIM 310200
Frequenza La Distrofia Muscolare di Duchenne (DMD) è una mala&a degenera,va della muscolatura con trasmissione X-‐linked recessiva, che colpisce 1 su 3.500-‐5.000 maschi na, vivi.
Fig.4 Southern blot su individui sani e affe& con rela,va dimensione della ripe,zione della tripleDa CGG.

Cause La distrofia muscolare è dovuta alla mutazione del gene DMD localizzato sul cromosoma X, che produce una grossa proteina chiamata distrofina, composta di 3.685 aminoacidi. La Distrofina è una proteina citoplasma,ca a forma di bastoncino che conneDe aDraverso la membrana il citoscheletro di una fibra muscolare con la matrice extra cellulare. In realtà forma un complesso proteico molto ar,colato di cui la distrofina è solo una parte (Fig. 1). La mancanza di distrofina induce la membrana cellulare a diventare permeabile ad alcune sostanze che di solito non potrebbero penetrare nella cellula, causandone l'esplosione e, quindi, la morte. A questo punto, il contenuto delle cellule morte viene riversato all'esterno, il sistema immunitario interviene efficientemente "ripulendo" una zona di muscolo più larga di quella danneggiata, lasciando un danno superiore a quello iniziale. Il "vuoto" creatosi nel muscolo viene riempito da tessuto conne&vo che provoca il successivo "strangolamento" delle cellule ancora sane. Tale processo si ripete in maniera costante fino alla morte di tuDe le cellule muscolari.
Mutazioni nel gene della distrofina causano distrofie di diversa severità, dalla forma grave come la distrofia muscolare di Duchenne (DMD; 310200)) a quella meno severa come la distrofia muscolare di Becker (BMD; 300376). Le analisi del DNA dei pazien, con DMD evidenziano delezioni che provocano un frame-‐shiz, ovvero uno sliDamento nella leDura del DNA, che produce un mRNA errato, per cui la distrofina non viene prodoDa o viene rapidamente degradata (Fig. 2).
Fig. 1 La Distrofina ha la funzione di conneDere la membrana al citoscheletro, rappresentato dai filamen, di Ac,na (in rosso) delle cellule del muscolo scheletrico e del muscolo cardiaco. La Distrofina (in giallo) è ancorata alla membrana sarcoplasma,ca mediante un complesso glicoproteico (β-‐distroglicano, Sarcoglicano, Sintrofina, Distrobrevina) agganciato alla matrice extracellulare.

Anche le mutazioni che causano la distrofia muscolare di Becker (BMD) sono prevalentemente (80-‐85% dei casi) grandi delezioni. Le delezioni però mantengono il modulo di leDura consentendo la produzione di una proteina più corta, ma semi-‐funzionale (Fig. 3). Circa 24% delle mutazioni sono even, de novo.
Manifestazioni cliniche Gli individui affe& vanno incontro a progressiva degenerazione della muscolatura; inizialmente viene interessata la muscolatura scheletrica, successivamente il difeDo di forza progredisce ulteriormente, coinvolgendo anche la muscolatura cardiaca e respiratoria. Non vengono invece interessa, i muscoli oculari estrinseci (quelli che consentono di muovere gli occhi) ed è poco coinvolta la muscolatura mimica del volto. L'esordio avviene nella prima infanzia e i bambini affe& possono presentare ritardo nelle tappe dello sviluppo motorio o ritardo globale. I bambini affe& da DMD non sono di solito capaci di correre o saltare. La mala&a ha un'evoluzione rapida
Fig. 2. a: DNA codificante la proteina funzionante. b: DNA con delezione che produce una proteina troncata all’estremità C-‐terminale e non funzionate
a
b
Fig. 3 Delezione di un grande traDo di DNA che porta ad una proteina più piccola ma parzialmente funzionante che causa la distrofia muscolare di Becker.

e il bambino sviluppa un'andatura anserina, caraDeris,ca andatura a papera, con segno di Gower posi,vo (Fig. 4) . Quest’ul,mo è il risultato di un test faDo al paziente sul modo di alzarsi da terra, si dice posi,vo se il paziente "si arrampica" su se stesso, puntellando gli ar, superiori e le ginocchia. I bambini con DMD hanno difficoltà a salire le scale e cadono frequentemente. La perdita della deambulazione autonoma avviene tra i 6 e i 13 anni; dopo la perdita della deambulazione, si sviluppano rapidamente le contraDure ar,colari e la scoliosi. La cardiomiopa,a e l'insufficienza respiratoria rappresentano la causa di morte dei pazien, all'inizio della vita adulta. Trasmissione A parte rare eccezioni, soltanto i maschi ne sono affe&, mentre le femmine portatrici sono in genere asintoma,che. Solo il 5% presentano sintomi che insorgono fra i 16 e i 48 anni e possono variare da una modesta generalizzata debolezza all’incapacità di movimento (ina&vazione sbilanciata del cromosoma X). Le femmine malate sono rare e manifestano la patologia di Becker. Vi sono anche casi nei quali le madri non sono portatrici e la mala&a è dovuta a una “nuova mutazione” avvenuta nella gametogenesi.
Diagnosi La diagnosi prenatale (tra la IX e l'XI se&mana) è possibile nelle famiglie nelle quali la diagnosi sia stata confermata con le analisi molecolari. È molto importante la consulenza gene,ca: il rischio di ricorrenza è del 50% nei fe, maschi di una madre portatrice. Le sorelle dei pazien, possono avere una probabilità del 50% di essere portatrici. La patologia è associata anche ad un’elevata concentrazione di crea,na-‐fosfo-‐chinasi (CPK) nel siero. L’enzima muscolare citoplasma,co esce dal muscolo perchè la sua membrana è degenerata. Il dosaggio elevato della CPK nel plasma può indicare femmine portatrici della mala&a (il valore della CPK in femmine portatrici è 100/200 volte superiore alla media).
Terapia e prognosi Per la ges,one della DMD è essenziale un approccio mul,disciplinare. La fisioterapia prevede lo stretching passivo e l'uso di ortesi caviglia-‐piede nelle ore noDurne per ridurre le contraDure del tendine di Achille. Il traDamento più efficace è l’uso di cor,costeroidi che servono a mantenere la massa e la forza muscolare. I cor,costeroidi devono essere introdo& quando lo sviluppo motorio del bambino inizia a rallentare, di solito tra i 5 e i 7 anni di vita; bisogna inoltre considerare le complicazioni dovute alla terapia con cor,costeroidi, controllando il peso, provvedendo alla somministrazione di farmaci per la protezione gastrica, garantendo il monitoraggio e il traDamento dell'osteoporosi e la visita oculis,ca, stante il rischio di cataraDa e di glaucoma. È necessario un regolare monitoraggio cardiaco per permeDere un traDamento tempes,vo con ACE-‐inibitori. Ques, inibiscono l’azione dell’enzima ACE (angiotensin conver,ng enzyme), che trasforma l’angiotensina I in angiotensina II, provocando la costrizione dei vasi sanguigni con conseguente
Fig. 4 Tipici movimenti di un paziente affetto da DMD per sollevarsi da terra.

aumento della pressione sanguigna. L’inibitore di ACE ha lo scopo di ridurre la formazione di angiotensina II, facendo si che i vasi sanguigni res,no dilata, e la pressione sanguigna diminuisca. La chirurgia può essere u,le per la correzione della scoliosi e la BIPAP noDurna (ven,lazione a due livelli di pressione posi,va intermiDente, Bi-‐level Posi,ve Airway Pressure) è efficace per il traDamento dell'insufficienza respiratoria restri&va. La DMD ha una prognosi sfavorevole e l'aspeDa,va di vita è significa,vamente ridoDa. Il decesso si verifica di solito all'inizio della vita adulta. La ges,one della DMD è in gran parte sintoma,ca: prevede la fornitura di disposi,vi di assistenza per camminare, la prevenzione della scoliosi e la pulizia delle vie respiratorie con vari metodi usa, per eliminare il muco e le secrezioni.
Due recen,ssime informazioni ritrovate nella leDeratura scien,fica vanno esaDamente in questa direzione. Ricercatori americani del Centro di Ingegneria Biomedica dell’Università Duke (Durham, Carolina del Nord, Sta, Uni,) sono riusci, a ricreare in laboratorio delle colture muscolari in 3D (hDps://elifesciences.org/content/4/e06430). Tali struDure (definite myobundle, ossia fasci muscolari) presentano le stesse capacità contra&li e una simile efficacia di risposta a vari s,moli eleDrici e farmacologici del muscolo umano, e sono un esempio di come una col,vazione di fibre muscolari alles,te in laboratorio possa rappresentare un modello per studiare la risposta alle varie mutazioni gene,che che causano la mala&a muscolare o, ancora più importante, l’eventuale efficacia terapeu,ca di possibili farmaci. In linea teorica, la strategia 3D potrebbe essere anche uno strumento per personalizzare le cure gene,che, sempre che le verifiche in laboratorio (già iniziate) siano incoraggian,.
Ricercatori giapponesi dell’Università di Kyoto, assieme a colleghi americani, hanno sviluppato una modalità di “correzione microchirurgica” del gene della distrofina u,lizzando cellule della pelle dei pazien,, trasformandole in cellule to,poten, (cellule staminali in grado di dare vita a un intero organismo e ad alcuni tessu, esterni necessari al suo sviluppo), ed u,lizzando un “bisturi molecolare” basato su una guida specifica che deriva dai baDeri (sistema CRISPR), per correggere il gene malato in modo mirato. La fase successiva degli esperimen, nel laboratorio di Kyoto ha permesso ai ricercatori di indirizzare le cellule to,poten, con il gene correDo a formare cellule muscolari e anche di verificare che la correzione permeDeva la produzione di distrofina normale e funzionante, senza apparentemente alterare altri geni (hDp://www.ncbi.nlm.nih.gov/pmc/ar,cles/PMC4297888/).
Sebbene per entrambe le nuove modalità di studio le ricadute sui pazien, siano ancora lontane e ci sia necessità di aDenta verifica sulla riproducibilità e sicurezza dei nuovi usi terapeu,ci offer, da queste metodologie, le recen, ricerche aprono nuovi orizzon, e nuove opportunità per la terapia della DMD.









![ManrinAmirullah,S.Ag.,ttrlA - staimaarif-jambi.ac.id · DAFTAR NII-AI UJIAI{ SEiiESTER GENAP STAI MA'ARIF JAIIBI TAHUN AKADE]TIIK 2A17 NOfi MATA KULIAH : Hukum Perbankan Syari'ah](https://static.fdocumente.com/doc/165x107/5cc4d83c88c993412c8cb439/manrinamirullahsagttrla-staimaarif-jambiacid-daftar-nii-ai-ujiai-seiiester.jpg)



![IL SEGNO LINGUISTICO VS. SEGNO MUSICALE NELLA VISIONE ... · „semn textual” [segno testuale], ce corespunde „semnului lingvistic” activ pe un alt palier al limbajului și](https://static.fdocumente.com/doc/165x107/5e2769a6fea98a0e760b9343/il-segno-linguistico-vs-segno-musicale-nella-visione-asemn-textuala-segno.jpg)



