
Ch.toxicologica
78
Ministerul Sănătăţii al Republicii Moldova Universitatea de Stat de Medicină şi Farmacie „Nicolae Testemiţanu” Facultatea Farmacie CATEDRA CHIMIE FARMACEUTICĂ ŞI TOXICOLOGICĂ TEMA :METODA CROMATOGRAFIEI GAZ-LICHIDE ŞI APLICAREA EI ÎN ANALIZA CHIMICO-TOXICOLOGICĂ. (Indicaţii metodice pentru studenţii anului IV)
-
Upload
igor-verdes -
Category
Documents
-
view
120 -
download
0
Transcript of Ch.toxicologica

Ministerul Sănătăţii al Republicii MoldovaUniversitatea de Stat de Medicină şi Farmacie „Nicolae Testemiţanu”
Facultatea Farmacie
CATEDRA CHIMIE FARMACEUTICĂ ŞI TOXICOLOGICĂ
TEMA :METODA CROMATOGRAFIEI GAZ-LICHIDE ŞI APLICAREA EI ÎN ANALIZA CHIMICO-TOXICOLOGICĂ.
(Indicaţii metodice pentru studenţii anului IV)
CHIŞINĂU 2009

Introducere În ultimele decenii în arsenalul preparatelor medicamentoase sau inclus un şir de preparate noi ca: sulfanilamidele, antibiotice, vitamine, antituberculoase, psihotrope, hormonale, cardiace ş.a. Creşterea arsenalului de forme medicamentoase este însoţit de apariţia noilor metode de analiză calitativă şi cantitativă. Aici nu este lipsită de mister metoda cromatografiei gaz-lichide.
Aceasta este, o metodă sensibilă, universală şi specifică, care oferă posibilităţi de identificare, dozare a substanţelor medicamentoase în preparate, lichide biologice, cât şi pentru separarea ingredientelor în amestec.
În prezent cromatografia gaz-lichide se aplică în lucrul practico-ştiinţific în laboratoarele biochimice, chimice, clinice ş.a. Pentru aplicarea acestei metode în practica chimico-farmaceutică nu sunt în de-ajuns surse în literatură.
Prin această indicaţie metodică oferim studenţilor un ghid de orientare în vederea analizei chimico-toxicologice a toxicilor volatili. Totodată sunt incluse metodele de îndeplinire a lucrărilor individuale, care ridică amplituda cunoştinţelor studenţilor în vederea determinarii substanţelor toxice din materialul biologic: sunt formulate scopurile, planul expus, materialul informativ, subiectele ce stau la baza studierii temei, indicaţii pentru lucrul practic al studenţilor în laborator.
Tema: Analiza toxicilor “volatili” prin metoda cromatografiei gaz-lichide
Scopul studierii temei: Ai învăţa pe studenţi să efectueze analiza chimico-toxicologica asupra toxicilor volatili din materialul biologic în urma izolării.
Planul studierei temeiI. Introducere în analiza cromatografiei gaz-lichide a toxicilor volatili.
Cercetarea materialului biologic asupra toxicilor volatili, care se izolează prin metoda antrenării cu vapori de apă.
II. Determinarea cantitativă a toxicilor volatili aplicând metoda cromatografiei gaz-lichide.
III. Determinarea alcoolului etilic în sânge şi urină prin metoda cromatografiei gaz-lichide.
Material informativEfectele cromatografice au fost cunoscute din cele mai vechi timpuri. Încă
pe timpul lui Aristotel oamenii dedurizau apa de mare, filtrând-o prin anumite pământuri.
Metoda cromatografică, ca metodă ştiinţifică de separare şi analiză a substanţelor, a fost elaborată de savantul rus M.S.Ţvet, în anul 1903. Cu ajutorul acestor metode Ţvet a stabilit, că pigmentul verde al plantelor clorofila, care se
1

consideră omogen, este într-adevăr alcătuit din câteva substanţe. Trecând extractul frunzei verzi printr-o coloană umplută cu praf de cretă şi spălându-l cu solvenţi organici Ţvet a obţinut câteva zone colorate, fapt care mărturisea incontestabil despre prezenţa în extract a câtorva substanţe. Ulterior acest fapt a fost confirmat şi de alţi cercetători. Această metodă a fost numită cromatografie (grec ”chromatos” - culoare) deşi însuşi autorul a demonstrat posibilitatea separării şi a substanţelor incolore.
În 1941 Martin şi Synge bazându-se pe fenomenul deja cunoscut ei încearcă separarea aminoacizilor, folosind o coloană umplută cu silicagel saturat cu apă, iar fază mobilă folosesc cloroform amestecat cu adausuri din ce în ce mai mari de alcool n-butilic. Astfel apare cromatografia de separare pe coloană.
În 1944 Gordon şi Martin pun bazele cromatografiei pe hârtie. Tehnica de lucru simplă face, ca cromatografia pe hârtie să se dezvolte şi să se aplice la separarea amestecurilor de substanţe organice şi ulterior la cele anorganice.
Datorită rapidităţii această metodă oferă informaţii printr-un efort minim.Cromatografia de gaze s-a dezvoltat datorită perfectării detectorilor,
folosirii diferitelor faze staţionare şi mobile noi.
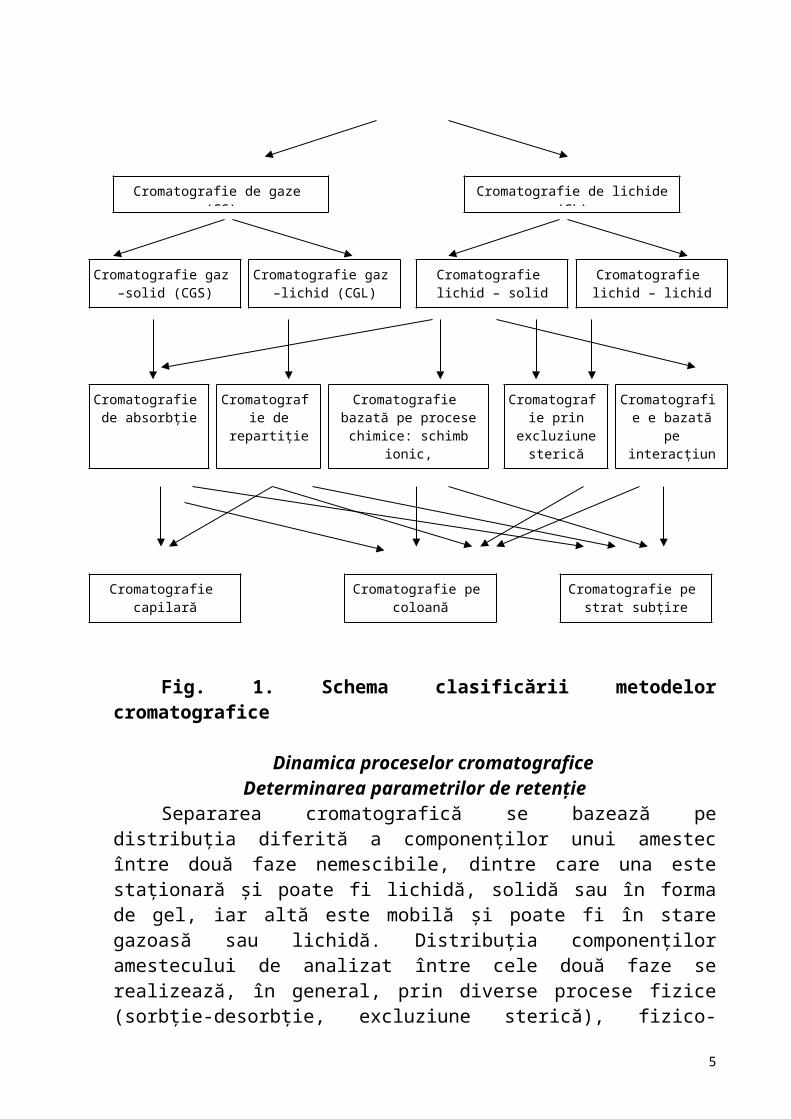
Clasificarea metodelor cromatograficeMetodele cromatografice de separare fac parte din metodele de separare,
bazate pe echilibrul între faze. În aceste metode este valorificată diferenţa între proprietăţile de separare a componenţilor amestecului între două faze de contact şi nemescibile. Clasificarea generală a metodei cromatografice este în funcţie de natura fazelor (I), procesele, care au loc la separare (II) şi modul de efectuare (III)(fig.1). Metodele de separare şi analiză cromatografică nu sunt specifice. În procesul de analiză cromatografică se ia în consideraţie natura fazelor, ce urmează a fi folosite, prezenţa compuşilor care interferă cu compuşii analizaţi. În alte cazuri este necesară izolarea unor compuşi necunoscuţi în vederea caracterizării lor ulterioare, sau analiza completă a unui amestec complex, necunoscut, prin separarea componenţilor, urmată de analiză de structură şi determinării cantitative.
Tehnicile de separare au fost create în funcţie de proprietăţile amestecurilor în limite extrem de mari), volatilitate şi alte proprietăţi.
Complexitatea tehnicilor de separare este corelată cu proprietăţile componenţilor de separat. În cazul izomerilor optici separarea lor poate fi realizată cu tehnici speciale. În alte cazuri proprietăţile sunt atât de diferite încât separarea poate fi realizată printr-o tehnică simplă (distilare).
După ce componenţii din amestec au fost separaţi se poate alege metoda adecvată de determinare.
2

Clasificarea metodelor cromatografice
Fig. 1. Schema clasificării metodelor cromatografice Dinamica proceselor cromatografice
Determinarea parametrilor de retenţieSepararea cromatografică se bazează pe distribuţia diferită a
componenţilor unui amestec între două faze nemescibile, dintre care una este staţionară şi poate fi lichidă, solidă sau în forma de gel, iar altă este mobilă şi poate fi în stare gazoasă sau lichidă. Distribuţia componenţilor amestecului de analizat între cele două faze se realizează, în general, prin diverse procese fizice (sorbţie-desorbţie, excluziune sterică), fizico-chimice (repartiţie, dizolvare-eliminare), chimice (schimb ionic, precipitare, complexare, oxido-reducere etc.). Cantitativ repartiţia componenţilor amestecului analizat, între cele două faze e determinată de raportul dintre cantitatea de component în faza staţionară şi mobilă şi se numeşte constantă de distribuţie a componentului în sistemul dat:
CS
K = (1)Cm
unde: K – constanta de distribuţie:
3
Cromatografie (C)
Cromatografie de gaze (CG) Cromatografie de lichide (CL)
Cromatografie gaz –solid (CGS)
Cromatografie gaz –lichid (CGL)
Cromatografie lichid – solid (CLS)
Cromatografie lichid – lichid (CLL)
Cromatografie de absorbţie
Cromatografie de repartiţie
Cromatografie bazată pe procese chimice: schimb
ionic, complexare, precipitare, oxido-
reducere
Cromatografie prin excluziune
sterică
Cromatografie e bazată pe
interacţiunea cu câmpul:
electroforeză
Cromatografie capilară Cromatografie pe coloană Cromatografie pe strat subţire

CS şi Cm – concentraţia (sau cantitatea) totală a tuturor formelor unui component analizat, prezent la echilibru în faza staţionară şi mobilă respectiv.
În funcţie de procesul predominant la separare, când componentul analizat e prezent numai într-o anumită formă în ambele faze deosebim constantă de repartiţie constanta de adsorbţie etc. În acest caz procesele secundare sunt neglijabile. Aceste constante se notează şi se calculează analogic constantei de distribuţie, iar CS şi Cm reprezintă concentraţia (sau cantitatea) de component într-o anumită formă în ambele faze.K – depinde de natura fazelor, t0 , pH, forţa ionică a soluţiei (dacă faza mobilă este lichidă) etc.
Substanţa este introdusă pe coloană cu faza mobilă. Ea se transportă (se reţine) în faza staţionară total sau parţial, fie prin adsorbţie, repartiţie, excluziune, reacţie chimică etc. Tot odată substanţa reţinută deja în faza staţionară contactează încontinuu cu noi porţii de fază mobilă şi total sau parţial se transferă din nou în faza mobilă. Faza mobilă deplasează încontinuu substanţele analizate pe un sector nou unde iarăşi din nou se realizează acelaşi act elementar de echilibru.
Fie că urmează să separăm, pe o coloană cromatografică, un amestec din doi componenţi A şi B. Cantitatea de substanţă în faza mobilă şi staşionară va fi corespunzător: [A]m, [B]m şi [A]S, [B]S. Din momentul introducerii amestecului pe coloană, la interfaţa dintre cele două faze nemescibile în conformitate cu legea echilibrului chimic, se stabilesc echilibre dinamice pentru fiecare component. Pentru fiecare component A şi B vom avea coeficientul de distribuţie KA şi KB, respectiv (fig.2).
Fig.2. Coloana cromatografică. Schema de formare a zonei de component în metoda de eluţie şi repartiyarea sub stanţei în zona de component
4
Cu cât componentul se reţine mai mult într-o fază cu atât şi concentraţia lui va fi mai mare în această fază şi mai mică în cealaltă fază, şi invers. De exemplu, dacă componentul A se reţine mai mult în faza staţionară decât componentul B, atunci, coeficientul respectiv de distribuţie vor îndeplini relaţia: KA > KB.
Orice separare cromatografică se caracterizează prin factorul de retenţie (sau retenţia) – R, care este raportul dintre viteza de migrare a componentului (VZ) şi viteza de migrare a fazei mobile (eluentului) – (V) în faza staţionară dată:
VZ
R = (2)V
Valoarea numerică a lui R este subunitară (0 < R < 1). În condiţii extreme, dacă VZ = 0, atunci R = 0 şi substanţa este totalmente reţinută pe coloana dată. Dacă VZ = V, atunci R = 1 şi substanţa este purtată de faza mobilă fără a interacţiona cu faza staţionară.
Retenţia depinde de coeficientul de distribuţie a substanţei date. Dacă R reprezintă fracţia moleculelor componentului ce se află în faza mobilă, atunci 1-R va reprezenta fracţia moleculelor aflate în faza staţionară la echilibru. Prin urmare se poate scrie:
CS 1 – R 1K = = → R = (3)
Cm R K + 1Din expresiile (2) şi (3) obţinem:
V VZ = (4)
K + 1De aici rezultă, că factorul de retenţie şi viteza de migrare a substanţei
prin coloană sunt invers proporţionale cu constanta de distribuţie a ei.Viteza de migrare a substanţei prin coloană poate fi definită ca raportul
dintre lungimea coloanei parcursă de substanţa dată (L) şi timpul de reţinere a substanţei (tR) pe coloana dată:
L VZ = (5)
tR
Luând în consideraţie expresia (4) obţinem: L (K+1)
tR = (6) V
Deoarece lungimea coloanei şi viteza fazei mobile în condiţiile date sunt mărimi constante, timpul de reţinere este direct proporţional cu constanta de distribuţie şi fiind uşor măsurabil, serveşte ca parametru de retenţie în analiza calitativă cromatografică şi se numeşte timp de retenţie.
5

Timpul de retenţie poate fi definit ca timpul în care substanţa dată a eluat (s-a deplasat) prin coloana de la momentul introducerii şi până la detecţia concentraţiei maxime la ieşirea ei din coloană.
Alt parametru de retenţie utilizat în cromatografie este volumul de retenţie (reţinere) total (V2), care poate fi definit ca volumul de eluent măsurat de la introducerea probei pe coloană şi până la ieşirea concentraţiei maxime a componentului de pe coloană.
Volumul de retenţie (VR) e proporţional cu timpul de retenţie (tR):
VR = tR F (7)unde F este viteza volumetrică a gazului purtător.
Parametrii de retenţii (tR, VR) depind de natura fazelor, de presiunea fazei mobile, masa fazei staţionare, pH, t0, parametrii geometrici ai coloanei etc. Dar asupra acestor mărimi pot influenţa şi diferiţi factori ocazionali.
Pentru componenţii A şi B parametrii de retenţie se vor afla în următoarele relaţii:
KA > KB
VA < VB
tR (A) > tR (B) (8)VR (A) > VR (B)
Prin urmare, componenţii amestecului analizat, având coeficienţi de distribuţie diferiţi se vor deplasa în procesul eluţiei cu diferite viteze şi vor avea un timp de reţinere diferit, în condiţiile date. În rezultat se va realiza separarea componenţilor pe coloana dată.
Constanta de distribuţie este factorul principal, care determină separarea substanţelor pe coloana cromatografică. Cu cât mai mare va fi diferenţa dintre valorile constantelor de distribuţie, cu atât mai efectivă va fi separarea cromatografică în condiţiile date.
Cromatografia poate fi definită ca un proces dinamic bazat pe realizarea multiplă a actelor de echilibru la distribuţia componenţilor amestecului în cele două faze nemescibile ce vin în contact.
În cromatografia de gaze separarea componentelor dintr-un amestec complex se realizează între o fază mobilă gazoasă şi o fază staţionară solidă (CGS) sau lichidă (CGL). Mecanismul separării poate fi un proces de adsorbţie (CGS) sau de repartiţie (CGL).
CG permite atât separarea substanţelor volatile (stabile la temperatura lor de volatilizare) cât şi a celor care în urma unor reacţii chimice (derivatizare) pot fi transformate în derivaţi volatili şi stabili.
Faza mobilă este constituită dintr-un gaz inert: He, Ar, Ne, H2, numit şi gazul vector (purtător), care antrenează substanţele de determinat.
Faza fixă (staţionară) este formată dintr-un adsorbent (CGS) sau dintr-un suport inert granulos (0,15-0,18 mm) impregnat cu un lichid de impregnare -
6

puţin volatil sau lichid selectiv (CGL). Faza fixă se găseşte tasată în coloană sau depusă pe pereţii unor coloane capilare. Aparatul şi principiile de lucru
În figura 3 este prezentată schema de princiăpiu a unui cromatograf de gaze de tipul „Crom-5”. Componentele esenţiale ale cromatografului cu gaz purtător, care are rolul de fază mobilă (1); reductor de presiune (2); uscător purificator (3); blocul de control fin (4); debitmetru (5); dispozitiv de injectare pentru introducerea probei (6); două coloane montate paralel (7); detectorul (8) şi imprimanta (11). Dispozitivul de injectare, coloanele şi detectorul sunt amplasate în cuptoare cu reglare şi control automatizat al temperaturii.
Fig. 3. Schema de principii a cromatografului de gaze Într-un cromatograf gazul vector care provine de la “sursa de gaz” reglat la un anumit debit de reglatorul de debit traversează camera de injectare a probei şi antrenează substanţele volatile spre coloana cromatografică. Volumul cuprins între capul de injectare şi capătul coloanei trebuie să fie cât mai mic posibil. O metodă mai nouă constă în injectarea direct în coloană, la 1-2 cm umplutură. Din coloană curentul de gaz este introdus în detector. Detectorul transformă diferenţa unei proprietăţi fizice dintre componentele probei şi gazul purtător într-un semnal electric, proporţional cu concentraţia componenţilor din faza gazoasă şi se produce un semnal, care se înregistrează cu ajutorul unui integrator, ordinator . Înregistratorul trasează o curbă în formă de clopot. Reprezentarea grafică a semnalelor detectorului în funcţie de timp, poartă denumire de cromatogramă (fig.4).
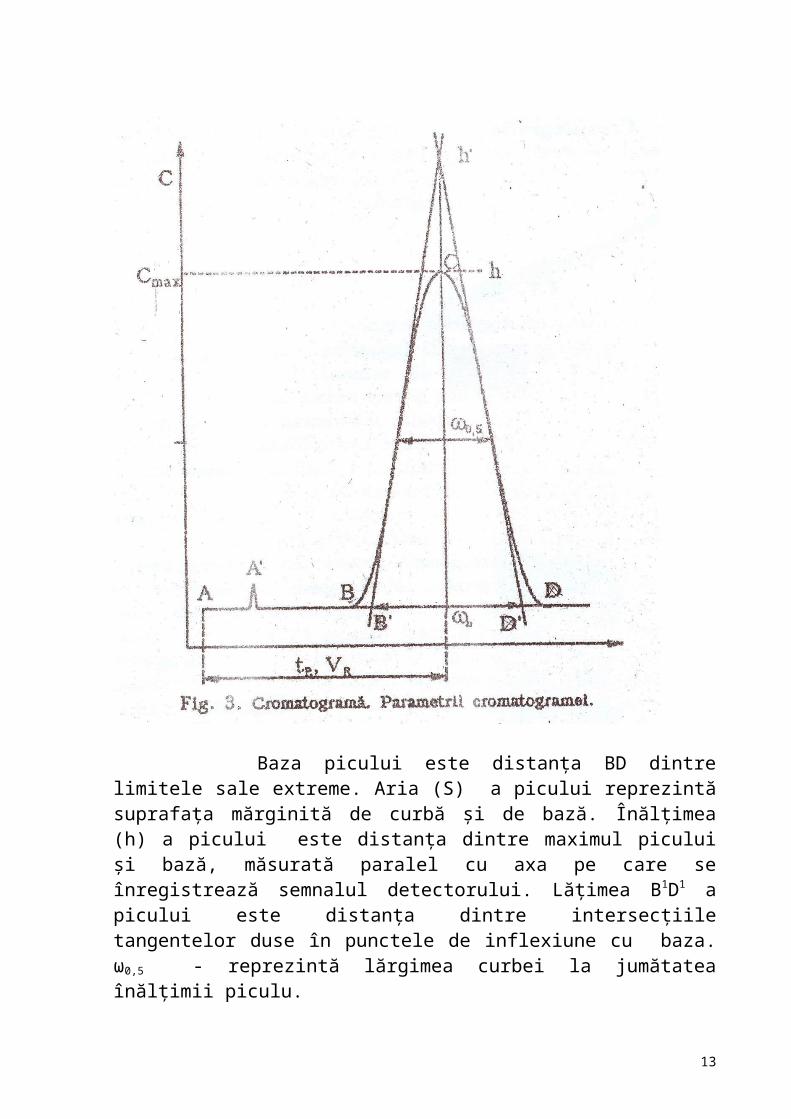
Picul cromatografic.Parametrii de bază şi mărimile derivate Picul este secţiunea cromatogramei semnalizată de detector la apariţia componentului în gazul purtător; în cazul separării incomplete, în acelaşi pic pot apărea mai multe componente. În gaz-cromatogramă se defineşte linia de bază a
7

cromatogramei – secţiunea de cromatogramă, care se înregistrează când din coloană iese numai gazul purtător.
Baza picului este distanţa BD dintre limitele sale extreme. Aria (S) a picului reprezintă suprafaţa mărginită de curbă şi de bază. Înălţimea (h) a picului este distanţa dintre maximul picului şi bază, măsurată paralel cu axa pe care se înregistrează semnalul detectorului. Lăţimea B1D1 a picului este distanţa dintre intersecţiile tangentelor duse în punctele de inflexiune cu baza. ω0,5 - reprezintă lărgimea curbei la jumătatea înălţimii piculu. Componentele probei se separă din coloană şi o părăsesc una după alta într-o anumită ordine, după intervale de timp determina
8

Fig.5. Parametrii de bază ai picului şi mărimile derivate
Timpul de retenţie (tR) – timpul aflării substanţei analizate în sistemul cromatografic. În practică se determină ca interval de timp din momentul introducerii probei în coloană şi momentul înregistrării amplitudinii maxime a picului cromatografic. Fiecare substanţă în unele şi aceleaşi condiţii îşi are timpul său de retenţie parametru ce stă la baza identificării componenţilor amestecului de analizat.
Timpul „mort” (to) – timpul de trecere a eluentului prin sistemul cromatografic prezintă mărimea, ce caracterizează un sistem cromatografic concret cu o viteză dată a fluxului de eluent. El se determină ca timp de eluare al componentului nereţinut.În practică determinarea timpului mort e dificilă, ce-i legată de inexistenţa substanţelor absolut nereţinute, din care cauză pînă în prezent nu-i elaborată o abordare unică de determinare. Astfel la determinarea to
prin metoda HPLC cu fază inversă se utilizează nitraţii, mai rar bromurile sau nitrometanul.
Timp de retenţie corectat sau net (recalculat) (tR) – timpul mediu, în decursul căruia moleculele substanţei se găsesc în faza staţionară. Se determină ca diferinţă: tR = tR – to. Factor de capacitate (coeficient de capacitate):K= tR/to=(tR- to)/to = tR/to– 1.Mărime proporţională coeficientului de distribuţie a sorbatului între faza mobilă şi staţionară. Este parametru mai universal, decât timpul de retenţie. Întrucât valoare lui nu depinde de demensiunile geometrice a coloanei şi de viteza de eluare. Cu mult mai rar se utilizează valoarea necorectată a factorului de capacitate: K = tR/to.
9

Înălţimea picului cromatografic (h) – distanţa dintre vârful picului şi linia de bază. Practic ea se determină ca lungimea perpendicularei, coborât din punctul de vârf al picului la linia de bază interpolară între capetele picului. Mărimea dată se utilizează la calcularea limitei de detecţie de asemenea în analiza cantitativă deopotrivă cu aria picului.
Lăţimea la jumătatea înălţimii picului (1/2) – distanţa orizontală dintre liniile, ce limitează profilul de pic la jumătatea înălţimii lui. Pentru picul gaussian 1/2 = 2,345 . Se utilizează la calcularea eficienţei coloanei şi altor mărimi.
Lăţimea la baza picului () se determină ca segment retezat la linia de bază de tangentele trasate în punctele de inflexiune pe pantele picului, sau ca distanţă orizontală dintre punctele situate la nivelul 0,044 h. Pentru picul gaussian = 4 . În practică este complicat de măsurat exact această valoare, de aceea mai des se utilizează mărimea 1/2.
Factor de asimetrie (As) – mărimea ce caracterizează gradul de asimetrie orizontală a picului. Pentru picurile simetrice, printre care şi gaussiene As=1; mărimea As 1 corespunde frontului ascendent mai întins a picului „fronting”, iar când As 1 este mai întins frontul descendent „tailing”. Există diverse procedee de determinare a factorului de asimetrie; cel mai des se aplică egalitatea: As = (a0,05 + b0,05) / 2a = 0,05/2a, unde a0,05 şi b0,05 – lăţimea corespunzător porţiunii anterioare şi posterioare a picului la nivelul 0,05 h.
Eficienţa coloanei (N) se exprimă prin numărul de talere teoretice şi este legată de parametrii funcţiei Gauss: N = tR/2.
Selectivitatea (factorul de separare) (a) capacitatea sistemului cromatografic de a separa doi analiţi.
a = tR2/tR1=(tR2- to)/tR1 - to = K2/ K1.
Ea reflectă doar gradul de separare a maximelor de pic fără a lua în consideraţie lăţimile lor.
Factorii care condiţionează separarea gaz-cromatografică sunt: 1. natura izotermei de repartiţie;2. volumul şi principiile de injectare în coloană a probei de analizat;3. natura gazului vector;4. programarea temperaturii şi a debitului de eluant;5. natura fazelor;6. tipul de detector. Forma picului cromatografic depinde de tipul izotermei de adsorbţie. La temperatură constantă adsorbţia se măreşte la creşterea presiunii gazului ori concentraţiei soluţiei.
Izoterma de adsorbţie este dependenţa cantităţii substanţei adsorbite de presiunea gazului purtărrtător sau concentraţia soluţiei la temperatură constantă. (fig.6)
10

Fig.6.Diferite tipuri de iz oterme
Aceste izoterme sunt descrise de o ecuaţie de tip L angmuir de forma: b c (9)
ai = am
1 + b cunde ai este cantitatea de substanţă reţinută (adsorbită) pe faza staţionară în timpul echilibrului; am – cantitatea maximă de substanţă care poate fi reţinută (adsorbită) pe faza staţionară dată, pentru acoperirea ei cu un strat monomolecular; b – constanta caracteristică puterii de adsorbţie; c – concentraţia componentului în faza mobilă.
După Langmuir la suprafaţa corpului solid există un număr de locuri cu energie minimă, aşezate la anumite intervale pe toată suprafaţa.Numărul lor este egal dm. Pe aceste locuri se pot adsorbi molecule de gaz ori din soluţie. În domeniul concentraţiilor mici izoterma e liniară (fig.6a). Pentru bc 1 numitorul din ecuaţia (9) devine egal cu 1 şi ecuaţia dată poate fi scrisă astfel:
ai = am bc = KHC (10)Această este ecuaţia adsorbţiei liniară. Corespunde ecuaţiei lui Henry (KH
– constanta lui Henry). Intervalul adsorbţiei liniare uneori se mai numeşte intervalul Henry. În cazul adsorbţiei liniare (fig. 6a) forma picului cromatografic este simetrică de tip goussian (fig.7a). În practică deseori dependenţa cantităţii substanţiei adsorbite de concentraţia soluţiei sau de presiunea gazului nu este liniară. Izoterma adsorbţiei poate fi de exemplu, curbată, de formă convexă (fig. 6b) şi atunci forma picului este asimetrică, cu coadă (fig.7b) sau de formă concavă (fig. 6c) şi atunci se obţin picuri „frontale” (fig. 7c).Izoterma de repartiţie trebuie să fie liniară pentru a obţine picuri cromatografice simetrice. În cazul izotermelor neliniare de adsorbţie se recomandă blocarea parţială a centrilor activi de adsorbţie prin depunerea unui lichid greu volatil sau să se lucreze la diluţii mari.
11
Fig.7 Forma picului în funcţie de tipul izotermei de adsorbţie

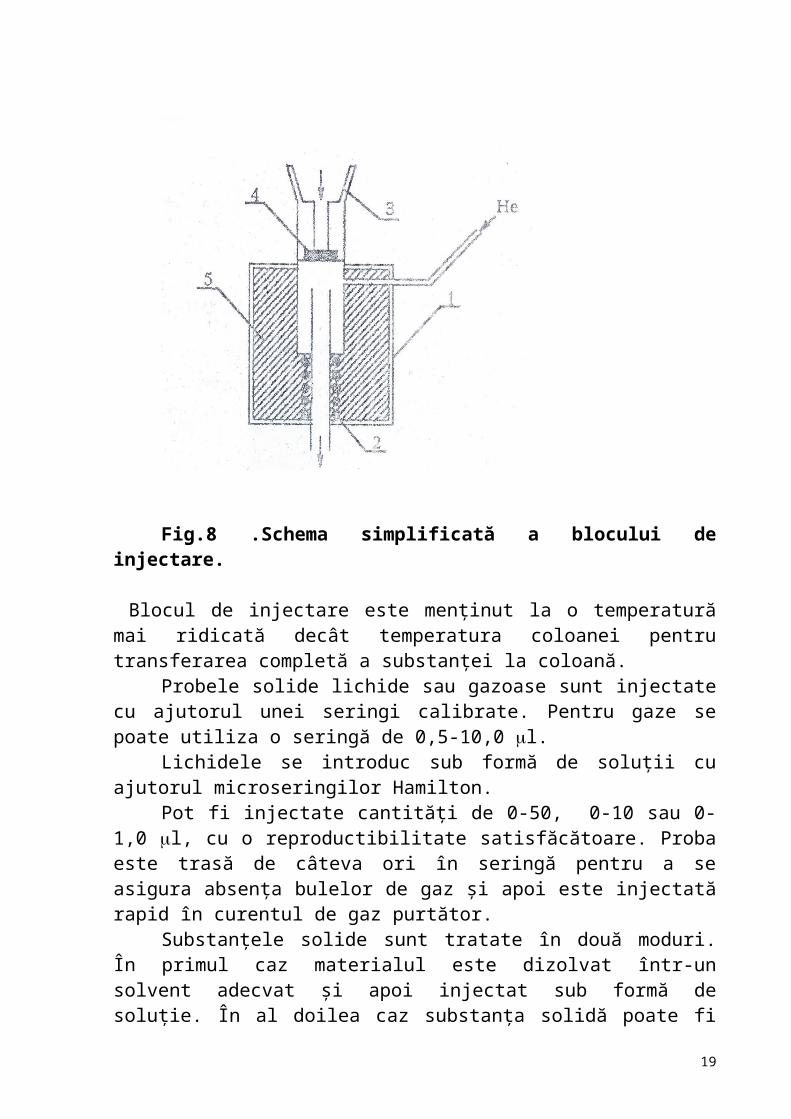
Dispozitivul pentru introducerea probei (fig.8) este amplasat, astfel, încât proba să fie introdusă direct în gazul de transport. El constă din corpul propriu-zis (1), elementul termic (2),radiato rmetalic(3), septum pliabil(4), prin care estinjectată proba.
Fig.8 .Schema simplificată a blocului de injectare. Blocul de injectare este menţinut la o temperatură mai ridicată decât temperatura coloanei pentru transferarea completă a substanţei la coloană.
Probele solide lichide sau gazoase sunt injectate cu ajutorul unei seringi calibrate. Pentru gaze se poate utiliza o seringă de 0,5-10,0 l.
Lichidele se introduc sub formă de soluţii cu ajutorul microseringilor Hamilton.
Pot fi injectate cantităţi de 0-50, 0-10 sau 0-1,0 l, cu o reproductibilitate satisfăcătoare. Proba este trasă de câteva ori în seringă pentru a se asigura absenţa bulelor de gaz şi apoi este injectată rapid în curentul de gaz purtător.
Substanţele solide sunt tratate în două moduri. În primul caz materialul este dizolvat într-un solvent adecvat şi apoi injectat sub formă de soluţie. În al doilea caz substanţa solidă poate fi injectată direct utilizând o seringă specială. Aceasta este concepută astfel ca substanţa să fie încărcată la capătul seringei printr-o „crestatură”. Proba este injectată, apoi printr-un septum, cu ajutorul unui plunger, direct în instalaţie. Dezavantajul acestui procedeu constă în lipsa de reproductibilitate, deoarece cantitatea de substanţă luată în seringă nu poate fi măsurată cu exactitate.
Probele care au presiuni de vapori scăzute sunt transformate pe cale chimică în derivaţi volatili, apoi injectaţi în instalaţie. Acesta este un procedeu
12
util, care să crească numărul compuşilor, care pot fi separaţi prin cromatografic de gaze. Ca urmare multe categorii de compuşi ca: aminoacizii, lipidele şi polimerii cu masa moleculară mare, care în mod normal nu sunt volatili pot fi separaţi după transformarea lor în derivaţi volatili. De exemplu acizii organici cu presiuni de vapori scăzute pot fi convertiţi în cloruri acide cu punctul de fierbere scăzut, steroizii pot fi silanizaţi sau ionii metalici pot fi complexaţi cu hexafluoroacetilacetonă. În toate cazurile presiunea de vapori a derivaţilor este mult mai ridicată decât presiunea de vapori a moleculei originale.Mărimea probei trebuie să corespundă concentraţiilor, care se încadrează în domeniul liniar al izotermei de repartiţie. Se pot determina probe gazoase, lichide şi solide. Gazele se introduc în injector prin prelucrarea lor de către eluant din dispozitivele speciale; substanţele lichide şi solide (după dizolvarea lor în solvenţi sau după topire) se introduc cu ajutorul unor microseringi sau capilare în volum de 0,1-10 ěl (concentraţia componentelor în probe fiind de ordinul mcg, mg ). Introducerea probei în injector se face rapid; temperatura injectorului trebuie să fie superioară p.f. al componenţilor, pentru a se realiza evaporarea instantanee a acestora.
Faza mobilă – gazul purtător. În mod obişnuit în cromatografia de gaze drept fază mobilă se foloseşte heliu sau azot. Aceste gaze sunt folosite în majoritatea cazurilor, deoarece îndeplinesc următoarele condiţii:
- faza mobilă trebuie să fie inertă- faza mobilă trebuie să aibă un preţ de cost redus, deoarece se folosesc
cantităţi mari.- faza mobilă trebuie să permită ca detectorul să răspundă într-un mod
adevărat. În calitate de rezervor pentru faza mobilă se foloseşte o butelia pentru
gaze rezistentă la presiune înaltă. La ea se ataşază un regulator de presiune pentru a reduce şi controla curgerea gazului prin coloană şi un debitmetru, pentru a controla debitul de gaz. Natura gazului vector are un rol însemnat, ea poate influenţa timpii retenţiei a componentelor sau, prin adsorbţia acestuia pe suprafaţa fazei staţionare, îi modifică proprietăţile. Alegerea eluantului se face în funcţie de sensibilitatea detectorului, de interacţiunea acestuia cu componentele de separat, cu faza staţionară. În cromatografia cu gradient de temperatură componentele se distribuie în coloană pe direcţia gradientului în funcţie de volatilitatea lor (cele mai puţin volatile – la intrare, cele mai volatile – la ieşire).
În cromatografia cu programare de temperatură o temperatură constantă pe toată lungimea coloanei variază în timp după o funcţie liniară sau neliniară.
Alegerea fazelor şi a suportului pentru un proces cromatografic se realizează în dependenţă de natura substanţelor care urmează a fi separate.
13

Dacă fazele sau suportul reacţionează cu compuşii injectaţi în coloană pot rezulta picurile de eluţie eronate, derive de fond ale detectorului sau chiar distrugerea instalaţiei.
Numărul de suporturi inerţi şi de faze lichide staţionare disponibile pentru CGL este practic nelimitat. Pentru a realiza o separare satisfăcătoare, faza lichidă aleasă trebuie să satisfacă următoarele condiţii.
- să fie un solvent bun pentru componenţii probei;- să posede o selectivitate sporită, pentru ca puterea sa de solvatare să fie
diferită pentru fiecare component al probei;- să posede presiune de vapori cât mai scăzută;- să fie stabilă din punct de vedere termic;- să fie inertă din punct de vedere chimic, faţă de componenţii probei
analizate.Criteriul cel mai important pentru alegerea fazei staţionare lichide este
polaritatea fazei şi a amestecului, care trebuie separat. Cea mai bună separare este realizată atunci când faza lichidă este similară din punct de vedere structural cu componentele amestecului, deoarece eficienţa separării depinde de presiunile de saturaţie a componentelor din amestec şi de coeficienţii de activitate a lor în raport cu faza dată. În cazul unor componente cu presiuni de saturaţie foarte apropiate, separarea va depinde în cea mai mare măsură de coeficienţii lor de activitate.
Coeficientul de activitate () reprezintă interacţiunile dintre moleculele componentului dizolvat în faza staţionară lichidă şi moleculele fazei staţionare. Aceste interacţiuni se manifestă prin forţe Kesson (între două molecule cu dipoli permanenţi), forţe de inducţie Debyl (între un dipol permanent şi unul indus, fie a fazei staţionare, fie a fazei mobile) şi forţe de dispersie sau forţe London (între moleculele nepolare), care se datoresc câmpului electric ai dipolilor cu viaţa foarte scurtă, produşi de mişcarea sistemului nucleu – electroni ai unei molecule.
Este avantajoasă folosirea umpluturilor, suprafaţa cărora este acoperită cu o peliculă subţire de fază staţionară lichidă. Însă micşorarea grosimii peliculei este limitată atât de interacţiunea componentului cu suportul fazei lichide prin fenomenul de adsorbţie, care se poate suprapune peste fenomenul de dizolvare, cât şi de necesitatea reducerii corespunzătoare a cantităţii de probă.
Fazele lichide pot fi clasificate în:- faze lichide nepolare: hidrocarburi lichide, uleiuri siliconice (cauciuc
siliconic, SE-30 etc.). Aceste faze separă componentele în ordinea creşterii temperaturii de fierbere.
- faze lichide polare conţin o cantitate mare de grupe polare (polietilenglicolii dimetilsulfolan etc.) Aceste faze separă numai compuşi polari.
- faze lichide cu polaritate intermediară cuprind fazele lichide cu grupe polare sau polarizabile pe un schelet mare nepolar (SE-52, dizodecilftalat, difenilbenzil etc.). Pe ele se separă compuşi cu polaritate intermediară.
14

- fazele lichide cu punţi de hidrogen sunt faze polare care conţin un număr mare de atomi de hidrogen capabili de a forma legături de hidrogen cu compuşii de separat.Caracterizarea fazelor lichide se poate face cu ajutorul constantelor lui Mc
Reynolds. Acest sistem se bazează pe cel al lui Rohrschnelder şi este mai potrivit pentru practica cromatografică.
Constantele Mc Reynolds (IR) sunt diferenţele între indicii de retenţie Kovacs (IR) pentru un component i pe faza lichidă de studiat şi faza de referinţă (IR
o).IR1 = IRi - IRi
o
S-a stabilit, că din toate fazele cunoscute, preferate sunt 12- Ele acoperă întreg domeniul de polarităţi. Aranjarea într-o scară a polarităţilor se face prin distanţa faţă de squalan, care se determină pe faza constantelor Mc Reynolds. În figura 9 este redată scara celor 12 faze preferate în cromatografia de gaze şi distanţele respective faţă de squalan.
2000 D1885 TCEF 1,2,3 – Tris (2 cianotoxi)propan1612 DEGS Dimetilglicolsuccinat
15001259 DEGA Dimetilglicoladipinat1052 PEG-20M Polietilenglicol
1000821 XE-60 Cianoetilmetil-siloxan709 GF-1 Trifluorpropilmetil-siloxan
500488 OV-22 Fenilmetildifenil-siloxan377 DC-610 Fenilmetil-siloxan271 OV-3100 SE-30 Dimetil-siloxan
00 Squalan
Fig. 9. Cele 12 faze staţionare lichide frecvent utilizate în CGL şi polaritatea lor în comporaţie cu squalanul . Faza staţionară solidă în CGS este constituită din adsorbenţi de tipul: silicagel, cărbune activ, site moleculare. Separarea se bazează pe un proces de
15

adsorbţie. Eficacitatea separării creşte prin impregnarea adsorbantului cu mici cantităţi de lichid nevolatili (siliconi) sau cu gaze care se adsorb ireversibil (CO2, H2O, H2S). Mai recent se utilizează coloane împlute cu polimeri organici cu porozitate mare, sub formă de perle, de exemplu din seria “Porapak” (etilvinilbenzen-divinilbenzen). Aceştia servesc în special la analiza apei, alcoolilor, gazelor cu masa moleculară mică şi amestecurilor lichide.
În scop analitic se utilizează coloane de 1-1,5 m lungime şi 3-6 mm diametru, care pot fi confecţionate din metal, sticlă sau material plastic şi au formă de U sau de spirală.
Dintre suporţii utilizaţi amintim cei de tip diatomit (Chromosorb P) Kieselgur (Chromosorb G,W, Sterchamol) polifluoretilena (Fluoropak-80, perle de sticlă). În cazul coloanelor capilare, rolul suportului îl joacă pereţii capilarelor, pe care se depune un strat subţire de suport solid, care ulterior va fi impregnat cu faza lichidă.
Capacitatea de separare a fazei staţionare lichide, în cazul amestecurilor depinde de raportul tensiunilor de vapori a componenţilor cât şi de tăria legăturilor ce apar între moleculele componentelor şi cele ale fazei staţionare. Deosebim legături ionice, Van der Waals, legături de hidrogen sau cu transfer de sarcină.
Pentru separarea componentelor cu puncte de fierbere foarte diferite, se pot utiliza combinaţii de două sau mai multe coloane în serie sau în paralel, cu un singur, respectiv cu doi detectori (ex. Coloana cu fază lichidă nepolară – Squalan şi una polară – Carbowax).
Detectori. În cromatografia de gaze, pentru depistarea componentelor, care ies de pe coloană se folosesc detectorii. În funcţie de proprietatea măsurată deosebim:
- detectori de ionizare (detectori de ionizare în flacără, detectori cu captură de electroni);
- detectori care măsoară o proprietate fizică de volum (detector de conductibilitate termică);
- detectori optici (flamfotometru, spectrofotometru);- detectori electrochimici. Clasificaţi în alt mod, detectorii pot fi: universali,
selectiv, specifici.Condiţiile generale pentru detectori sunt: sensibilitatea, stabilitatea,
fiabilitatea şi emiterea unui semnal liniar pentru anumit domeniu de concentraţie a probei.
Detectorul de conductibilitate termică (DCT) sau catarometrul are cel mai răspândit domeniu de utilizare (fig10). El face parte din categoria detectorilor universali. Este un detector nedestructiv şi poate fi utilizat şi în cromatografia preparativă.
16
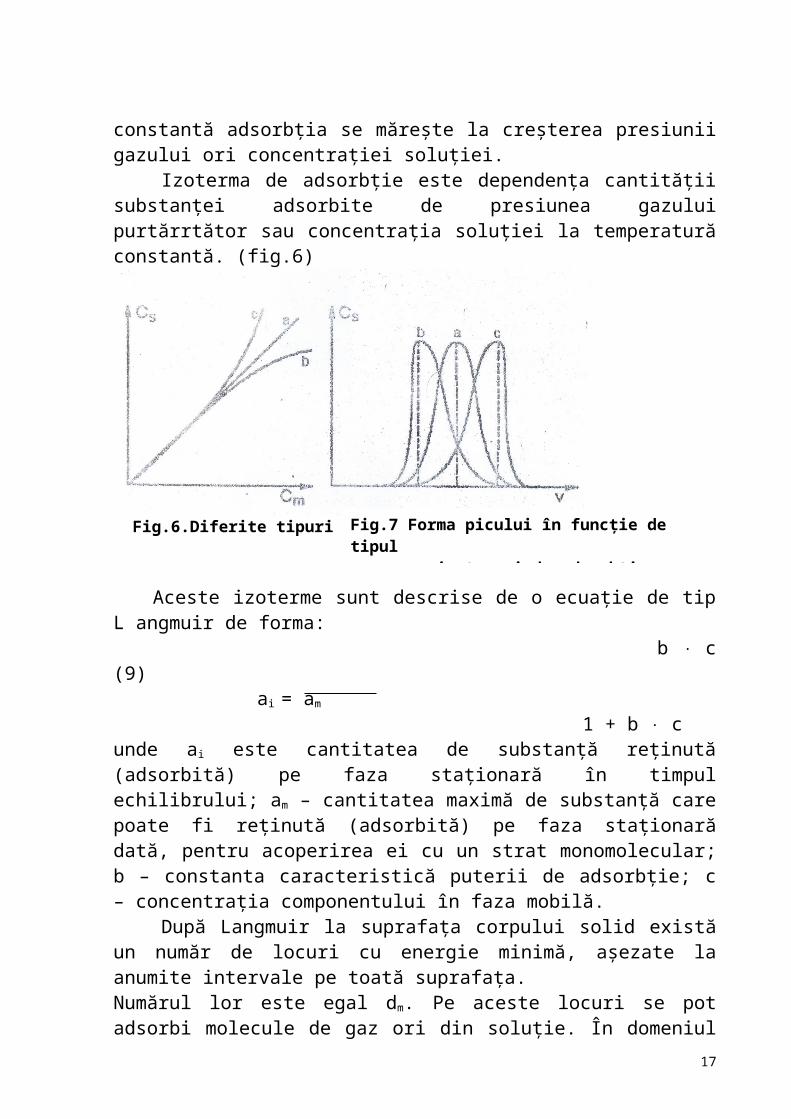
Fig. 10. Schema de principiu a unui detector cu conductibilitate termică (DCT)
Principiul de funcţionare se bazează pe măsurarea diferenţei de conductibilitate termică între component şi eluent. În acest scop se foloseşte un montaj electronic, cu două sau patru celule de conductibilitate termică. În figura 12 este redată schema unui montaj cu patru celule. Două din celule sunt spălate continuu cu gaz purtător (R1 şi R4 – celule de referinţă), celelalte două sunt spălate de amestecul binar care iese din coloană (R2 şi R3 – celule de măsură). Constructiv celulele nu pot fi realizate identice. Pentru echilibrarea iniţială a punţii se foloseşte potenţiometrul 4. Filamentele detectorului sunt confecţionate dintr-un material a cărui rezistenţă electrică variază foarte mult în funcţie de temperatură. În timpul funcţionării, filamentele sunt încălzite de trecerea unui curent continuu stabilizat, de la o sursă de curent 1. Stabilirea curentului de lucru se face cu un potenţiometru 2 şi se măsoară cu miliampermetrul 3.
Temperatura detectorului, în procesul de lucru trebuie să fie mai înaltă decât temperatura coloanei. Atât timp cât la ieşirea din coloana cromatografică va apărea numai gazul purtător, rezistenţele celulelor vor fi aceleaşi, puntea va fi echilibrat şi instrumentul de măsură montat va indica 0.
În registrătorul 5 va fi trasată linia zero. Prezenţa unui component în eluentul, care circulă prin celula de măsură modifică conductibilitatea termică a mediului gazos din această celulă. În rezultat se modifică rezistenţa electrică a filamentelor R2 şi R3, care provoacă dezichilibrul punţii. Intensitatea curentului înregistrat la dezichilibrarea punţii este proporţională cu concentraţia componentului care iese din coloană.
Mărimea semnalului detectorului depinde de diferinţa dintre conductibilitatea termică a componentului şi eluentului. Conductibilitatea termică a substanţelor depinde de masa moleculară ei. Hidrogenul, heliul şi azotul, având mase moleculare mici posedă conductibilităţi termice ridicate. Aceste gaze sunt ideale pentru a fi folosite ca gaze purtătoare în cromatografia de gaze.
17

Sensibilitatea detectorului depinde de factorul geometric al celulelor (dimensiunile, raza, lungimea filamentului) şi de valoarea curentului de alimentare a punţii. Sensibilitatea creşte la mărirea curentului în punte.
Creşterea curentului peste o anumită valoare poate duce la distrugerea filamentelor şi de aceea valoarea lui este limitată de natura gazului purtător şi de temperatura de lucru a detectorului.
Linia zero este sensibilă la variaţiile debitului de gaz purtător şi al temperaturii Ppentru ca variaţiile temperaturii să fie cât mai mici blocul metalic al detectorului trebuie să aibă o masă cât mai mare, iar celulele să fie dispuse simetric în detectori.
Detectorul de ionizare în flacără (DIF) este un detector universal, stabil şi simplu în construcţie. Principiul de funcţionare a detectorului cu ionizare în flacără de hidrogen constă în măsurarea conductibilităţii electrice a unei flăcări de hidrogen în prezenţa unui compus organic (fig. 11).
Fig. 11. Schema detectorului cu ionizare în flacără de hidrogen
La ieşirea din coloană eluentul este amestecat cu un curent de hidrogen şi este aprins la capătul unei duze din platină sau oţel inoxidabil. Duza este montată într-o cameră de ardere alimentată cu un curent de aer sau oxigen. De asupra flăcării este montat un electrod colector (anod). Alimentarea electrozilor se face de la o sursă de tensiune continuă, de valoare mare (100-350 V). La arderea H2
în prezenţa numai a gazului purtător între duză şi electrodul colector se stabileşte un curent foarte slab (10-14 A).
Apariţia în flacără a unui compus organic măreşte intensitatea curentului (datorită ionizării acestuia) care poate să ajungă până la 10-5 – 10-8A. în funcţie de natura şi concentraţia componentului respectiv. Acest curent va fi amplicat şi înregistrat.
Sensibilitatea detectorului se defineşte ca fiind cantitatea de sarcină electronică, produsă de un gram de component prin ionizare în detectorul cu flacără. Sensibilitatea detectorului pe mol de substanţă este proporţională cu
18
numărul atomilor de carbon din moleculă legaţi numai de hidrogen sau alţi atomi de carbon din moleculă plus contribuţia atomilor de carbon legaţi de halogeni, amine sau grupe hidroxil. Atomii de carbon oxidaţi complet din moleculă (din grupa carboxil sau carbonil) nu au nici o contribuţie la valoare sensibilităţii.
Detectorul de ionizare în flacără de hidrogen nu poate detecta gazele permanente (N2, H2, He, Xe), oxizii de azot, compuşii, care conţin un atom de carbon singur legat de oxigen sau sulf (CO2, Cs2, COS) gaze anorganice (NH3, SO2), apă şi acid formic. Limita de detecţie constituie 10-13 g.
Determinări calitative Aprecierea calitativă a unei gaz-cromatograme care prezintă un număr de
picuri se face în mod obişnuit prin:1) examinare vizuală, în comparaţie cu gaz-cromatograma unei probe etalon;2) compararea timpului (tR) sau a volumului de reţinere (VR) a probei de
determinat cu a etalonului-pur, cromatografiat în condiţii identice, sau cu datele menţionate în literatură pentru compusul respectiv;
3) compararea cromatogramei amestecului de determinat cu cromatograma aceluiaşi amestec, la care s-a adăugat substanţa etalon care se bănuieşte a fi prezentă în probă. Dacă picul unui component apare mărit pe a doua cromatogramă, acesta este un indiciu al prezenţei, în amestec, a substanţei adăugate probei; în caz contrar, substanţa adăugată dă un pic nou pe cromatograma a doua;
4) identificarea componentelor se mai poate realiza cu ajutorul detectorilor specifici; DCE se utilizează pentru identificarea compuşilor cu afinităţi diferite pentru electroni (alcooli, eteri, esteri, derivaţi carbonici, derivaţi halogenaţi). Spectrometrul de masă înregistrează spectrul de masă (SM) al componentelor prezente în fracţiunea de eluant introdusă în sursa de ioni a spectrometrului, cu înregistrarea concomitentă a cromatogramelor obţinute prin trecerea celeilalte fracţiuni printr-un detector universal (CT);
5) identificarea prin sorbţie sau reacţie selectivă se realizează prin intercalarea între injector şi coloana de separare a GC a unei coloane umplute cu adsorbant, sau absorbant specific unei anumite clase de substanţe, sau a unui microreactor în care are loc transformarea sau reţinerea prin reacţie chimică a unui component.
Timpul de retenţie (reţinere) brut (tR), exprimat în secunde sau minute, este timpul care se scurge între momentul introducerii probei în injector şi timpul când picul atinge maximul.
Timpul relativ (tR) este timpul de retenţie al unui component (i) în raport cu al unui component etalon (e) : tRi / tRe.
Timpul de retenţie corectat este dat de diferenţa dintre timpul de retenţie brut (tR) şi timpul la care se separă eluantul sau un component nereţinut pe coloană (ideal) şi al componentului respectiv: Rx= tR /tRx. Factorul de retenţie
19

are valori cuprinse între 0,01-0,9, în cazul unei separări optime, când valoarea se apropie de 1, înseamnă că acel component X, părăseşte coloana odată cu eluantul.
De aici se defineşte şi factorul de separare, pentru doi componenţi A şi B:tR(A)
(AB) = --------- , care trebuie să aibă valori ≠ 1 (11)tR(B)
Eficacitatea coloaneiO importanţă deosebită în prezentarea teoretică a procesului
cromatografic de separare o au două teorii. Teoria „Talerului teoretic” şi teoria cinetică.
Teoria „talerului teoretic” a fost propusă de Martin şi Synge. Conform acestei teorii coloana cromatografică se împarte imaginar într-un şir de segmente elementare – talere – şi se presupune, că pe fiecare taler se realizează un act elimentar de echilibru la distribuţia substanţei în faza staţionară şi faza mobilă. Fiecare porţie nouă de fază mobilă purtătoare (gaz sau lichid) provoacă o deplasare a acestui echilibru, în urma căruia o parte de substanţă se deplasează pe următorul taler, pe care la rândul său, se stabileşte un nou echilibru distributiv şi are loc trecerea substanţei pe următorul taler. În urma acestor procese substanţa supusă cromatografiei se distribuie pe câteva talere. Concentraţia substanţei pe talere din mijloc este maximă în comparaţie cu talerele invecitate.
Talerul teoretic poate fi definit ca un segment imaginar din coloana cromatografică în care se realizează un echilibru elementar complet de distribuţie a componentului dintre cele două faze nemiscibile, care vin în contact în procesul separării cromatografice.
Talerul teoretic din coloana cromatografică este analogic talerului teoretic dintr-o coloană pentru distilare fracţionată. Deosebirea constă în aceea, că componenţa amestecului, care ese de pe coloana cromatografică încontinuu se schimbă dar în procesul de extracţie ori distilare se poate de obţinut pe parcursul întregului proces una şi aceeaşi substanţă.
Numărul talerelor teoretice pentru componentul dat şi pentru lungimea coloanei date diferă de numărul talerelor teoretice pentru alţi componenţi în aceleaşi condiţii. Numărul talerelor teoretice este direct proporţional cu lungimea şi diametrul coloanei.
Numărul de talere N este un număr adimensional şi se numeşte eficienţa coloanei cromatografice, H şi n – parametrii teoretici, care caracterizează eficacitatea de separare a coloanei cromatografice şi determină dispersia zonelor de component.
Cu cât coloana va avea mai multe talere teoretice pe o unitate de lungime şi cu cât înălţimea echivalentă unui taler teoretic va fi mai mică, cu atât mai
20

efectivă va fi coloana pentru separare în condiţiile date. Aşa dar, condiţiile eficacităţii de separare pe coloana cromatografică sunt :H să obţină valori cât mai mici şi N – cât mai mari.
Dacă vom raporta lungimea stratului de fază staţionară (L) în care s-a produs separarea amestecului de substanţe la înălţimea talerului teoretic, vom obţine numărul de talere teoretice (N) necesare pentru separarea amestecului dat de coloana dată (formula 12).
LN=
HTeoria talerelor teoretice presupune realizarea procesului cromatografic în
trepte, dar în realitate procesul decurge incontinuu. Această teorie ne permite să se calculeze parametrului cantitativi importanţi pentru caracterizarea procesului cromatografic de separare. Aceşti parametri îşi menţin importanţa şi în teoria cinetică a cromatografiei, care ţine cont de viteza de migrare a substanţei, difuzie şi alţi factori cinetici, care caracterizează cantitatea procesului.
Teoria cinetică a cromatografiei acordă o atenţie fundamentală cineticii procesului, asociind înălţimea echivalentă talerului teoretic (H) cu procesele difuziei, stabilirea lentă a procesului.
Parametrul experimental care determină din punct de vedere dinamic eficacitatea separării unei coloane cromatografice este lăţimea picului cromatografic, numită în literatură şi lărgirea zonei. Zona îngustă şi compactă a componentului de la începutul coloanei (la introducerea probei) se va lărgi, astfel încât concentraţia pe unitate de volum de coloană se va micşora. (fig.2).
Lărgirea zonei acţionează în sensul micşorării separării, ducând la o reamestecare a componenţilor, respectiv la o suprapunere a picurilor cromatografice. Pentru practica cromatografică este necesară cunoaşterea tuturor factorilor, care influenţează dispersia zonei în vederea acţionării asupra lor în sensul obţinerii unor picuri cât mai înguste.
Giddings a elaborat teoria privind lărgirea zonei (), moleculelor în coloană e considerată ca un proces haotic incontinuu. Principalii factori, care duc la lărgirea zonei unui component ce parcurge coloana sunt:
a) difuzia moleculară longitudinală;b) cinetica sorbţie-desorbţie;c) transferul de masă în faza mobilă (format din difuzia transversală şi
structurală sau turbulentă.Prin însumarea acestor contribuţii se obţine înălţimea echivalentă talerului
teoretic. În funcţie de viteza fazei mobile, care într-o formă simplificată e cunoscută sub denumirea de ecuaţia lui Van Deemeter:
BH = A + + C V (13)
Vunde: ABC – constante: V – viteza fazei mobile.
21

Această ecuaţie se aplică în cazul cromatografiei cu gaze. Contribuţia fiecărui termen al ecuaţiei (13) asupra înălţimii echivalente talerului teoretice (H) în funcţie de viteza fazei mobile (V) e ilustrată în fig.12.
Fig.12. Reprezentarea grafică a ecuaţiei Van Deemter Constanta A e în legătură cu acţiunea difuziei de vârtej, care depinde de mărimea particulelor şi de densitatea fazei staţionare în coloana dată. Mărimea B e legată cu coeficientul de difuzie a moleculelor în fază mobilă şi ia consideraţie acţiunea difuziei longitudinale. Mărimea C caracterizează cinetica procesului de sorbţie-desorbţie, transferul de masă şi alte efecte. Primul termen difuzează un aport constant în H. Efectul celui de-al doilea termen e esenţial pentru o viteză mică a fluxului. Cu mărirea vitezei fazei mobile creşte influenţa celui de-al treilea termen, iar a celui de-al doilea se micşorează.
Curba rezultantă H = f(v) se caracterizează printr-o valoare minimă a înălţimii echivalente talerului teoretic (H min), care corespunde unei viteze optime a gazului purtător (v opt).
Pentru a determina acest punct, diferenţiem ecuaţia (13) şi egalăm cu derivata ei cu zero:
dH B= - + C = 0 (14)
dV V2
obţinem V opt = B/C. Înlocuind acestă mărime în ecuaţia (13) determinăm înălţimea echivalentă a talerului teoretic minimă (Hmin) la care separarea cromatografică va fi cel mai efectivă:
Hmin = A + 2 BC (15)Astfel teoria cinetică oferă bază pentru optimizarea procesului
cromatografic.
22

Factorii, care influenţează eficacitatea de separare:a) Viteza fazei mobile. Influenţa este redată prin ecuaţia lui Van Deemter.b) Mărimea şi suprafaţa specifică a particulelor fazei staţionare.
Numărul de talere creşte odată cu micşorarea dimensiunii particulelor suprafeţei specifice. Dimensiunile particulelor nu pot fi prea mici, deoarece creşte brusc rezistenţa opusă de coloană la trecerea fazei mobile.
În literatura de specialitate există tabele în care se specifică relaţiile optime dintre diametrul coloanei şi mărimra particulelor fazei staţionare.
c) Natura fazelor. Alegerea fazelor în dependenţă de natura probei ce urmează a fi analizată. Cantitatea de fază staţionară, luată pentru separare trebuie optimizată. În cazul cromatografiei gaz-lichid de exemplu, o cantitate prea mare de fază staţionară lichidă face ca particulele de umplutută să devină lipicioase şi să se aglomereze, afectând astfel, eficacitatea de separare a coloanei şi condiţionează apariţia unor picuri cu cozi. Micşorarea cantităţii de fază staţionară lichidă măreşte eficacitatea coloanei dar o cantitate prea mică va spori interacţiunea fazei purtătoare cu particulele suportului.
d) Parametrii geometrici a coloanei (lungimea, diametrul, forma). Din acest punct de vedere coloanele capilare sunt cele mai eficiente. Creşterea lungimii şi micşorarea diametrului coloanei conduce la creşterea numărului de talere. Dar totuş există o lungime şi un diametru optim limitat de fenomenele de curgere, care provoacă dispepsia zonelor (tendinţa de reamestecare).
e) Temperatura. În cazul cromatografiei de repartiţie (gaz-lichidă, mărirea temperaturii măreşte separarea, dar în cromatografia de adsorbţie mărirea temperaturii influenţează negativ separarea.
Eficacitatea coloanei se măsoară cu numărul talerelor teoretice. Pentru compararea eficacităţii coloanelor trebuie de identificat faza staţionară, substanţa de analizat, temperatura, viteza gazului vector şi mărimea probei.
Numărul talelor teoretice (N) se determină după formua:.
X 2
N = 16 y
în care:X – este distanţa din momentul introducerii probei până la maximul picului substanţei de analizat, mmy – Distanţa formată de linia de bază a picului cu tangentele duse în punctele de inflexiune ale picului cu baza, mm.
23

2. Înălţimea echivalentă unui taler teoretic (H): L
H = N
unde:L – lungimea coloanei cromatografice în cm;N – numărul de talere teoretice.
3.De trasat curba dependenţei H (pe ordonată) de viteza gazului purtător (pe abcisă).De determinat condiţiile optime.De completat tabelul:
VHe X y X/y (X/y)2 N=16(x/y)2 H
Calcularea reproductibilităţii experimentului prin metoda gaz-cromatografică.
Fiecare student primeşte 3 cromatograme a unei substanţe, descrie condiţiile experimentului, măsoară înălţimea picurilor în mm.
Eroarea unei determinări:hmediu – h1 = a1
hmediu – h2 = a2
hmediu – h3 = a3
Eroarea medie: a1 + a2 + a3
= b 3
Eroarea medie relativă : b˙ 100
hmediu
Eroarea relativă medie: b / hmediu∙ 100. Eroarea relativă medie nu trebuie să fie mai mare de 3-4 %.
6) Derivatizarea sau transformarea prealabilă a componentelor polare (greu volatile sau care se descompun la temperatura lor de fierbere) în derivaţi mai puţin polari a permis separarea şi identificarea omologilor aceleiaşi clase, a
24

izomerilor etc. Astfel, substanţele cu grupe polare (OH, COOH, NH2) se pot transforma în compuşi cu grupe mai puţin polare (OCOR şi NHCOR).
Analiza cantitativă în cromatografie se bazează pe stabilirea unei relaţiei dintre mărimea semnalului dat de detector şi cantitatea de compus prezent în probă. Semnalul detectorului este măsurat prin înălţimea sau aria picului. Aria picurilor poate fi calculată conform relaţiilor).
s = ½ h0 s = ½ h0
s = ½ h0,5 s = 2,507 h Aria picului poate fi măsurată cu ajutorul planimetrului, poate fi apreciată
după greutatea fâşiilor de hârtie tăiate după conturul picurilor. În prezent în acest scop se folosesc integratoare electronice şi microcomputere, care înregistrează foarte rapid şi exact orice mărime de pe cromatogramă.
Metodele principale în analiza cromatografică cantitativă sunt: a) metoda standardului intern, În probă cu o compoziţie cantitaţivă necunoscută se introduce preventiv o cantitate exactă de substanţă cunoscută, care nu se conţine în amestecul dat, nu interacţionează chimic cu componentele amestecului şi este stabilă la temperatura la care se efectuează analiza. Proprietăţile fizico-chimice ale substanţei adăugate (standardului intern) trebuie să fie asemănătoare proprietăţilor componentelor amestecului cercetat. Partea de masă (XI) a fiecărui component (în %) se determină din relaţia:
Si rXi = 100
Sst
unde Si şi Sst sunt ariile picurilor componentului analizat şi standardului, corespunzător; r – raportul masei standardului intern către masa probei: masa standardului
r = masa amestecului de analizat fără standard
b) metoda normării ariilor. Această metodă presupune, că suma tuturor ariilor picurilor corespunzătoare componentelor amestecului cercetat constituie 100 %. Partea de masă (Xi) a componentului i (in %) se determină din următoarea relaţie:
Si
Xi = n=i 100 % (41) Si
c) metoda etalonării absolute. Se prepară şi se cercetează o serie de soluţii standard. Se determină experimental dependenţa înălţimii sau suprafeţei picului
25

de concentraţia substanţei şi se trasează curba de etalonare. Apoi se determină aceeaşi parametri ai picurilor ai amestecului de analizat şi din curba de etalonare se determină concentraţia substanţei analizate. Această metodă simplă şi exactă este principală metodă de determinare a microimpurităţilor. Metoda nu necesită o separare a tuturor componenţilor amestecului, dar se limitează la analiza componentului studiat în amestecul concret.
. La baza determinărilor cantitative stă relaţia de proporţionalitate între aria de sub picul cromatografic şi cantitatea componentului eluat. Cheia acestor metode constă în măsurarea ariei şi stabilirea acestei proporţionalităţi.
1) Măsurarea ariei se poate face pe mai multe căi:- Măsurarea ariilor este înlocuită uneori cu măsurarea integrală a înălţimii
picului (h), sau a jumătăţii ei (h/2) (figf. 7).- În cazul picurilor simetrice aria se măsoară prin metoda produsului dintre
lăţimea picului (determinată la ˝ înălţimii picului) şi înălţimea lui (perpendiculară coborâtă din vârf pe linia de bază) (fig.7) sau din aria triunghiului format de linia de bază cu tangentele duse în punctele de inflexiune ale picului:
h ∙ y A = --------
2- Aria este proporţională cu masa picului decupat şi cântărit, cu condiţia ca
grosimea hârtiei să fie uniformă.- Ariile se mai pot determina automat, utilizând în acest scop un integrator
mecanic sau un computer.2) Corelarea datelor obţinute cu compoziţia cantitativă a probei. Dintre
metodele care stabilesc relaţiile pentru transformarea şi corelarea corectă a ariilor cu concentraţia componentelor, amintim:
a) Metoda standardului extern: se realizează într-un mod asemănător curbei de etalonare din spectroscopie. Cantităţi cunoscute de probă-etalon se cromatografiază; se măsoară ariile corespunzătoare picurilor şi se întocmeşte curba de etalonare, care are înregistrată în ordonată valoarea ariilor (mm2) şi în abscisă concentraţia.(g).
b) Metoda standardului intern constă în introducerea unui standard, (substanţă de referinţă) în cantitate cunoscută, în proba de analizat. Drept standard intern se poate utiliza o substanţă pură, care să se elueze cu o rezoluţie bună, cu un timp de reţinere apropiat de al componentelor de determinat şi care să nu interacţioneze cu acestea. Cunoscând concentraţia standardului intern în probă (Cs) şi măsurând ariile picurilor corespunzătoare componentei de determinat (Ax) şi ale standardului (As) se poate calcula concentraţia necunoscută (Cx). În acest scop se întocmeşte o curbă de etalonare sau se calculează factorul de corecţie al ariilor pentru compusul de determinat (fx).
26

Curba de etalonare se realizează prin cromatografierea a trei soluţii de concentraţii exacte din componenta de determinat (C1, C2, C3), prin dizolvarea ei în soluţia standardului intern (Cs). Se determină, în fiecare caz în parte, raportul ariilor: A1/As; A2/As; A3/As. Se înscriu aceste valori în ordonata şi concentraţiile C1, C2, C3 în abscisa unui sistem de coordonate şi se trasează curba de etalonare.
Factorul de corecţie al ariilor (fx) se poate calcula astfel:Se face media aritmetică (Vx) a valorilor rapoartelor obţinute mai sus:
V1 + V2 + V3 A1/As = V1; A2/As = V2; A3/As = V3; ---------------- = Vx
3Pentru fiecare concentraţie (C1, C2, C3) se calculează raportul: C1/Vx;
C2/Vx; C3/Vx; Media aritmetică a valorilor obţinute pentru aceste trei concentraţii, reprezintă factorul de corecţie mediu, fx, care intervine în formula de calcul.
Cx (mg/ml) = fx • Vx
c) Metoda normării ariilor. Această metodă se bazează pe faptul că raportul existent între aria corespunzătoare picului unui anumit component SA şi suma ariilor tuturor componentelor prezente (∑ S) este egală cu procentul de component prezent în amestec. SA
A % = --------------------- ∙ 100. SA + SB + SC + ...
Relaţia de mai sus este valabilă numai în cazul în care sensibilitatea detectorului este aceeaşi pentru toate componentele probei, în caz contrar se recomandă ca etalonarea să se facă în prealabil cu ajutorul unor factori de corecţie.
Concentraţia se calculă după curba de etalonare ori după formula:Setilnitrit
C = ------------ ∙ F ∙ R ∙ 100,Spropilnitrit
în care:C – concentraţiaS – ariile corespunzătoare a picurilorF – factorul de corecţie a ariilor ori a înălţimii picurilor, ce se determină în
raport cu standardul în condiţiile experimentului (faza staţionară, to injectorului, to termostatului, detectorului; viteza gazului vector)
F – (factorul) se calculă după formula:
Sst
F = ------ ∙ C %; 'în care: Sx
27

Sst – aria picului standarduluiSx – aria picului substanţei de analizat
ori hst
F = ------ ∙ C %; în carehx
Sst – înălţimea picului standarduluiSx – înălţimea picului substanţei de analizat.C % - concentraţia substanţei de analizat.
Se calculă F pentru câteva concentraţii şi apoi se i-a media: masa standardului
R = -----------------------------------------------------masa amestecului de analizat fără standard.
Identificarea alcoolului etilic prin metoda gaz-cromatograficăa) În băuturile alcoolice şi soluţii alcoolice. Metoda constă în aceea, că
alcoolii sunt transformaţi în alchilnitriţi, care se separă în coloana cromatografică.
Pentru aceasta într-un flacon de sub penicilină se introduce 0,5 ml soluţie de acid tricloracetic 50% şi 0,5 ml soluţie de alcool etilic cu concentraţie exactă în limitele 3-4% (soluţie standard). Flaconul se închide cu dopul de gumă, care este fixat de un dispozitiv cu un orificiu în dop. Cu ajutorul seringii în flacon prin dop se introduce 0,25 ml soluţie de nitrit de sodiu 30%. Conţinutul flaconului timp de un minut se agită. Cu altă seringă uscată se i-a 3 ml din faza gazoasă ce conţine etilnitrit şi se injectează în cromatograf. După obţinerea cromatogramei soluţiei standard se determină timpul de retenţie corectat.
Apoi tot aşa se cromatografiază soluţia de analizat şi se compară timpul de retenţie brut, corectat cu a soluţiei standard. Coinciderea confirmă identitatea lor.
În sânge şi urină e asemănător cu identificarea etanolului în băuturi alcoolice şi soluţii alcoolice. Mai întâi se cromatografiază şi se determină timpul de retenţie a soluţiei etanolice (standard). Apoi se determină etanolul în sânge şi urină. În flacon de sub penicilină se introduce 0,5 ml sânge ori urină şi 0,5 ml soluţie 50% de acid tricloracetic, se închide cu dop şi se fixează cu fixatorul.
Apoi se introduce prin dop 0,25 ml soluţie de nitrit de sodiu 30%. Conţinutul flaconului bine se agită timp de un minut. Apoi din flacon cu o seringă curată se i-a 3 ml fază gazoasă, se introduce în cromatograf şi se cormatografiază. Dacă coincide timpul de retenţie a soluţiei standard şi a substanţei ce se conţine în sânge ori urină, atunci se confirmă identitatea lor. Lucrarea N 1Tema: Introducere în cromatografia gaz-lichidă a toxicilor volatili. Cercetarea
substanţelor toxice care se izolează din materialul biologic prin metoda antrenării cu vapori de apă.
28

Scopul lucrării: Studierea metodelor de identificare a toxicilor volatili aplicând metoda cromatografiei gaz- lichide.
Planul 1. Controlul cunoştinţelor practice.2. Lucrul de sinestătător asupra identificării toxicilor volatili prin metoda CGL.3. Concluziile lucrării şi controlul deprinderilor practice.4. Oformarea proceselor verbale.
Subiectele ce stau la baza pregătirii teoretice1. Însuşirea tehnicii de lucru cu cromatograful: a regula parametrii de bază –
temperatura, viteza gazului vector, a controla lucrul principiilor de dozare.2. Însuşirea tehnicii de analiză a probelor lichide şi gazoase cu conţinut de
substanţe toxice.3. Prelucrarea cromatogramelor, calculând timpul de retenţie corectat şi relativ.4. Identificarea toxicilor volatili prin metoda cromatografiei gaz-lichide.5. Principiile de bază ale CGL.6. Schema cromatografului gaz-lichid. 7. Camera de evaporare şi funcţiile ei. 8. Coloanele cromatogafice şi rolul lor în separarea substanţelor. Faza
staţionară şi cerinţele faţă de ea.9. Tipurile de detectori şi sensibilitatea lor.10.Noţiune de cromatogramă şi pic cromatografic.11.Timpul de retenţie în coloană.12.Metodele de determinare calitativă în analiza cromatografiei gaz-lichide.13.Factorii care acţionează asupra separării substanţelor în coloana
cromatografică eficacitatea coloanei, selectivitatea fazei staţionare.14.Etapele de bază în identificarea toxicilor volatili izolaţi din materialul
biologic prin metoda CGL.1. Ce factori acţionează la separarea componentelor în coloana CGL?2. Explicaţi noţiunea de eficacitate a coloanei cromatografice.3. Care sunt parametrii ce caracterizează înălţimea echivalentă talerelor
teoretice?4. Cerinţele, care se atribuie către fază lichidă staţională.5. Definiţia coeficientului de repartiţie componentelor în cercetare prin metoda
CGL.6. Natura formelor de interacţiune între substanţele de cercetare şi faza
staţionară.7. Reciprocitatea dintre coeficientul de repartiţie şi forţele de interacţiune a
componentului cu faza staţionară lichidă.8. Care sunt parametrii de care depinde timpul de reţinere şi corectat în coloana
cromatografului?
29

9. Principiile de bază pentru identificarea toxicilor volatili în lichidul biologic prin metoda CGL.
.Răspundeţi argumentat la următoarele întrebări
1. În ce compartiment al cromatografului se efectuează separarea substanţelor.2. Care este prioritatea metodei CGL faţă de metodele chimice pentru analiza
toxicilor “volatili”.3. Cum înţelegeţi definiţia “înălţime”, “talere teoretice echivalente”.4. Ce dozatore se folosesc pentru transmiterea probelor gazoase şi lichide în
coloana cromatografului?5. Cum se schimbă mărirea tipului de retenţie a substanţelor în coloană în
timpul analizei calitative?a. De la momentul introducerii probei.b. De la maximum picului de aer.c. De la maximum picului vecin?
6. Ce este timpul de retenţie corectat şi timpul de retenţie relativ şi de ce parametrii ai cromatografului depind ei?
1. Cum se va petrece repartiţia pe coloana cromatografică, care conţine 20 % glicerină a următorilor amestecuri?
Metanol, etanol, propanol, butanolPropanol, benzenOctan, benzen
2. Argumentaţi răspunsurile la aceleaşi întrebări dacă în calitate de sorbent ar fi 20 % ulei de vaselină.
3. Daţi explicaţie expresiilor:- timpul reţinerii- distanţa de reţinere- volumul de reţinere
4. Prin ce se determină selectarea fazei staţionare lichide?5. Cu ajutorul cărei faze staţionare lichide ПЕG-1500 ori Triton X-100 este mai
eficientă rfepartiţia amestecurilor eterilor metilici ai acizilor graşi? Argumentaţi răspunsul.
Ce proprietate a substanţelor de cercetat determină cantitatea necesară a bazei lichide staţionare în coloana cromatografică? Arătaţi aproximativ cantitatea fazei lichide staţionare pentru repartiţia amestecurilor hidrocarburilor aromatice, alcoolilor alifatici, substanţe medicamentoase
Răspundeţi în scris la următoarele întrebări
Problema 1. Pentru separarea substanţelor folosirea ca detector în caterometru se folosesc diferite gaze-vectori- azot, heliu, hidrogen, argon. Ce gaz-vector asigură o sensibilitate mai mare a catarometrului. Argumentaţi răspunsul alcătuind următoarea tabelă.
30

Gaz Conductibilitate termică
M. Mol
Problema 2. În secţia chimico-judiciară e necesar de determinat cantitatea metanolului în urină prin metoda cromatografiei gaz-lichide. Ce detector trebuie să folosim, ca la determinarea metanolului să nu împiedice apa?Problema 3. În secţia chimico-judiciară trebuie de efectuat analiza-express a materialului biologic asupra microcantităţilor de cloroform în sânge. Ce tip de detector trebuie să folosim pentru soluţionarea acestei probleme?
Problema 4. Cetăţeanul A fost internat în centrul pentru tratarea intoxicaţiilor acute cu presupunere de intoxicare cu fenol. A fost efectuată analiza gaz-cromatografică a urinei acidulate a pacientului. Pe coloana polară timpul relativ de reţinere corespundea etilenglicolului şi fenolului, iar pe cea nepolară fenolului şi toluolului. Ce concluzii a făcut chimistul-expert?Problema 5. Pentru identificarea picului cromatografic se folosesc următoarele caracteristici:a. Punctul iniţial al piculuib. Maximul piculuic. Lăţimea piculuid. Înălţimea picului.e. Suprafaţa picului.
Lucrul practic al studenţilor în cadrul lucrării de laboratorStudenţii fac cunoştinţă cu tehnica de securitate.
Lucrul practic constă din 2 etape:I. Determinarea timpului de reţinere relativ a substanţelor pure pe coloane
de polaritate diferită şi completarea tabelei generale.II. Identificarea toxicilor “volatili” în amestecul-model.
1. Fiecare student primeşte 1 substanţă o cromatografiază pe coloane cu polaritate diferită. Se prelucrează cromatogramele obţinute.
2. Fiecare student primeşte amestecul model, conţinând 2 substanţe necunoscute şi le cromatografiază pe ambele coloane cu polaritate diferită în aceleaşi condiţii.Cromatogramele se prelucrează, calculând timpul de reţinere corectat şi relativ. Apoi identificarea se efectuază după schema aplicată în cercetarea cromatogramelor model.
.3 Determinarea timpului de retenţie relativ a componentelor pure pe coloana cu fază staţionară lichidă de polaritate diferită şi alcătuirea tabelei generale. (Lucrarea I).
4. Identificarea toxicilor volatili în amestecul model (Lucrarea II).
31

Aparatura şi detaliile aucziliare1. Cromatograful gaz-lichid cu detector şi termostat la t 100 şi m.m.2. Microsering pentru 1mcl şi 10mcl.3. Flaconul cu lichide pentru spălarea microseringelor: cu alcool, cu eter.4. Linia de măsurare, triunghi, lupa. 5. Colba cu apă de săpun şi periuţa Regulile pentru studenţi la folosirea microseringilor1. La sfârşitul lucrului seringa se spală cu alcool, eter, luând fiecare solvent în
seringă de 2-3 ori şi eliberând pe hârtie de filtru.2. Seringa se aranjază în penal.3. Se interzice de a injecta în coloană acul până a fi colectată proba.4. Nu se admite colectarea rapidă a probei.
Pregătirea către analiza cromatografică1. Înainte de a începe lucrul verificăm ermeticitatea camerei de evaporare.
Pentru aceasta cu ajutorul periei şi soluţiei de săpun se unge deschizătura evaporatorului, unde ar trebui să troducem acul siringei. Ermeticitatea se constată prin lipsa vaporilor de gaz în această regiune.
Controlul ermeticităţii se efectuază şi în timpul lucrării.2. Controlul eficacităţii siringii de 1 l se face prin colectarea 0,5 l acetonă şi
dozarea lui pe hârtia de filtru. În timpul dozării apare o micropicătură pe vârful acului, iar la atingerea ei pe hârtie de filtru – apare un spot umed. Dacă picătura lipseşte atunci se întăreşte acul de siringă.În procesul lucrului ermeticitatea microseringii se controlează de fiecare dată
după 7-10 probe.Îndeplinirea analizei cromatografice
Analiza gaz-cromatografică începe cu aranjarea bandei de autoânscriere. Apoi în microseringă se colectează 0,5l solvent organic şi se introduce în camera de evaporare a cromatografului. Se înseamnă cu creionul momentul introducerii probei. După aceasta se înregistrează picul aerului, care se înseamnă prin “Aer”, după care urmează să fie picul probei de analizat compusului standard ori amestecul standard propus de profesor. Analiza aerului şi la toate celelalte probe se interpretează consecutiv nu mai puţin de 2 ori pe aceeaşi coloană. Astfel se controlează suprapunerea cromatogramelor paralele după timpul de reţinere, adică după distanţa de la momentul introducerii până la apariţia maximului şi după mărimea înălţimii lui. Dacă rezultatele obţinute de pe ambele cromatograme nu se suprapun se repetă experienţa până la suprapunerea rezultatelor. În cazuri contrare calitatea cromatogramelor paralele se analizează cu profesorul.
Analiza substanţelor lichide se îndeplineşte cu ajutorul microseringei de 1l ori 10 l .Mărimea probei 1-0,5 l. Sensibilitatea pentru fiecare substanţă se alege individual. După îndeplinirea analizelor paralele seringa se spală de câteva ori cu soluţia de analizat şi numai atunci este gata de colectat proba pentru îndeplinirea experienţei următoare pentru aceeaş probă.
32

Analiza cromatogramelorIniţial se măsoară cu ajutorul riglei distanţa (mm) de la momentul
introducerii probei până la maximul picului de aer (fig.1). Această distanţă corespunde timpului de găsire a componentului în interiorul cromatografului în stare mobilă.
Mărimea T este distanţa de reţinere a gazului neabsorbit. Se măsoară ea cu ajutorul riglei în mm. Distanţa obţinută în mm raportată la viteza mişcării corespunde timpului de reţinere a gazului neabsorbit în sec ori min.
Distanţa de la maximul picului aerului până la maximul picului compuşilor de analizat, raportat la viteza lentei corespunde timpului de retenţie corectatat al amestecului de analizat (T). Dacă nu se menţionează “ştrih” atunci este timpul de reţinere a gazului neabsorbit, care nu e adecvat pentru calcule. Pentru calculele de identificare se foloseşte numai timpul corectat (T), aşa cum numai el determină timpul, în care moleculele substanţei de analizat a “toxicului volatil” se găsesc în stare de absorbţie (dizolvare) în fază staţionară.
Numai timpul staţionării este diferit pentru fiecare toxic “volatil”, după cum numai el caracterizează solubilitatea diferită a toxicilor în una şi aceeaşi fază staţionară, ori absorbţia diferită in unul şi acelaşi absorbent.
Datele valorilor L şi T a fiecărui component se înscriu în tabelă Tabelul 1
Denumirea compusului Coloana cu sorbentul polar
Coloana cu sorbent polar
L : Tsec : Trel L : Tsec : Trel
- Cloroform- Alcool propilic- Etanol
Lx
TR = ----------------------
C (viteza lentei)Valoarea Trel se calculează cu exactitate până la 0,01. După efectuarea
calculelor şi îndeplinirea tabelului în procesul verbal studentul înscrie o tabelă o singură pagină, care conţine date despre condiţiile cromatografice a tuturor substanţelor cercetate.
33

Fig.1. Determinarea distanţei de reţinere pe cromatogramăa – momentul introducerii probei; b – maximum picului de aer;c – maximum picului substanţei de cercetat;Lx – distanţa de reţinere totalăLx – distanţa cercetată de reţinereL – distanţa de reţinere a gazului neabsorbit
Obiectele de cercetare- amestec model din 2 toxici volatili din următoarele componente:
cloroform, alcool – metilic, etilic, butilic, izobutilic, aceton, etilacetat, toluen, dioxan, benzen, tetraclorură de carbon, dicloretan, hexan. Etapele de lucru:
I. Determinarea timpului de retenţie relativ a componentelor pure pe coloana cu fază staţionară lichidă de polaritate diferită şi alcătuirea tabelei generale. (Lucrarea I).
II. Identificarea toxicilor volatili în amestecul model (Lucrarea II).Fiecare student primeşte amestecul model conţinând 2 substanţe
necunoscute şi efectuează cromatografierea pe două coloane de polaritate diferită în aceleaşi condiţii ce se referă la substanţele pure. (Lucrarea I).
Cromatogramele se prelucrează, calculând timpul de retenţie corectat şi relativ. Apoi identificarea se efectuează după schema dată. Pe cromatogramă se arată parametrii, care se folosesc pentru identificare.
Indicaţii pentru efectuarea identităţii toxicelor “volatili”Identificarea toxicilor “volatili” prin metoda cromatografiei gaz lichide se
reduce la Trel pe 2 coloane cu polaritate diferită. Dacă componentul nu se separă pe o coloană sub formă de pic, numaidecât separarea are loc pe a II coloană. Deaceea analiza amestecurilor se efectuează pe două coloane cu sorbent de polaritate diferită. Pe cromatogramele obţinute se calculează timpul de retenţie relativ pentru fiecare pic cromatografic. Parametrii obţinuţi se aranjază în două tabele. În prima tabelă se includ parametrii de reţinere pe coloana cu sorbentul polar, a doua – pe coloana cu sorbent nepolar. Aici se finisează I etapă de lucru.
În etapa II se folosesc datele individuale din tabelă obţinute pentru toxicii “volatili” În tabelul 2 includ parametrii de reţinere şi denumirile
34

corespunzătoare, la care timpul de retenţie relativ nu se deosebeşte decât cu 0,05, determinat experimental prin analiza amestecurilor de toxici neconoscuţi.
Exemplu de tabelă. Datele experimentale şi tabelare despre parametrii de reţinere a toxicilor “volatili” necunoscuţi pe coloana cu sorbent polar. Calculele timpului relativ de reţinere este îndeplinit în raport cu benzenul (ori alt solvent).
Tabelul 2Datele despre parametrii de reţinere a toxicilor “volatili” necunoscuţi pe coloana cu sorbentul polar
Nr pic pe cromatogramă
Datele experimentale Datele din tabelă
Determinarea substanţei
L (mm) Trel Trel
1 63 0,84 0,790,810,85
ButilformiatPropoxibutanMetanol
2 125 0,99 1,001,001,011,03
BenzenButilizobutiratCloroformDibutoxibutan
Datele din tabele despre parametrii de reţinere şi determinarea toxicilor “volatili”, care pot asista în problema de control e necesar să se continuă în tabela completată după rezultatele determinării timpului de reţinere relativ a toxicilor pure – substanţe de comparare – pe două coloane cu polaritate diferită. Aceste tabele sunt rezultatele lucrului practic a lucrării precedente.
Pentru etapa următoare lucrul constă în compararea tabelelor completate pentru coloanele cu diferită polaritate conţinând date concrete timpul de reţinere a toxicilor “volatili” necunoscuţi din problema de control şi denumirile propuse. De aici rezultată, că componentele care au pic comun pe o coloană să fie în aceeaşi măsură orientate şi pe altă coloană.
Aceste condiţii se respectă.Dar cu toate acestea sunt abateri, când necătînd la polaritatea diferită,
ambele coloane nu sunt selective pentru perechea dată de substanţe. În acest caz aceste substanţe urmează să fie excluse din ambele tabele şi necesitatea identificării lor se rezolvă separat după cum în amestecul de analizat ar putea să existe în mod separat ori împreună. În rezultat ambele componente se înscriu în răspunsul problemei “convenţional identificate”.
După excludere se funcţionează compararea tabelelor. Aceste denumiri corespund toxicilor, care persistă în obiectul de cercetare. Pentru finalizarea acestei etape de lucru urmează să completăm tabela finală, forma căreia este dată mai jos.
Tabelul 3Denumirea toxicilor “volatili” determinaţi pe fiecare
coloană cu polaritate diferităDenumirea toxicului
identificat
35

Coloana polară Coloana nepolară
Prima rubrică a tabelei se completează cu toate denumirile, găsite pe coloana polară, a II cu denumirile de pe coloana nepolară. Denumirile subliniate se înscriu în rubrica II. Acestea vor fi la rândul lor denumirea toxicilor cercetaţi.
Aşa dar, în procesul identificării, efectuat pe coloane paralele, denumirea toxicului, care se conţine în probă e necesar, ca să fie determinat după totalitatea parametrilor de repartiţie obţinuţi pe fiecare coloană folosită. Dacă denumirea toxicului e găsită numai după datele unei coloane, acest toxic în probă de cercetat nu se conţine. Unii şi aceeaşi toxici pe diferite coloane pot fi repartizaţi în ordine diferită.
Dacă pe cromatograme se înregistrează numai un pic şi nu este exclusă prezenţa lui în probă în calitate de toxic “volatil” apare necesitatea de a schimba benzenul cu un alt solvent de exemplu: butilacetat, alcool propilic. Controlul total1. Aprecierea rezultatelor obţinute în urma efectuării lucrului practic.2. Semnarea proceselor verbale. Bibliografie1. Materialul prelegerilor 2. Бабилев Ф.В., Тряпицына Т.П. Газожидкостная хроматография в
фармацевтическом анализе. Кишинев, “Штиинца”, 1978, р. 4-10, 15-19. 20-25. 30-53.
3. В.Ф. Крамаренко « Токсикологическая химия», Киев, Выща школа, 1989
4. Г.Мак. Нейр, Э.Бопелли. Введение в газожидкостную хроматографию. М., Мир, 1970. Р. 29-39.
5. Вигдергаиз М.С. Качественный газохроматографический анализ, гл. 3, М., “Химия”, 1979, р. 356.
6. Н.Перцев. Н. Корцев. Справочник по газовой хроматографии. Москва, “Мир”, 1987, р.246.
Lucrarea N2Tema: Metoda cromatografiei gaz-lichide în analiza chimico-toxicologică.
Determinarea cantitativă.
36

Scopul lucrării: A însuşi metodele de determinare cantitativă a toxicilor “volatili” prin metoda CGL. A studia teoretic şi a asimila practic metodica de analiză a CGL asupra alcoolul etilic în sânge şi urină aplicată în laboratoarele Centrului de medicină legală, şi laboratoarele chimice.
Planul 1. Controlul cunoştinţelor teoretice.2. Lucrul de sinestătător asupra metodei de determinare calitativă şi cantitativă
a alcoolului etilic în lichidele biologice.3. Concluziile lucrării şi controlul deprinderilor practice.4. Oformarea proceselor verbale.
Subiectele ce stau la baza studierei temei:1. Bazele teoretice ale CGL.
- eficacitatea coloanei cromatografice şi fazei staţionare lichide- selectivitate fazei staţionare- detectorii cromatografului
2. Determinarea cantitativă prin metoda cromatografiei gaz-lichide.3. Metodele de calculare a suprafeţei picurilor:
- normării a ariilor - standardului intern- calibrării absolute
1. Ce reacţie stă la bază identificării şi dozării alcoolului etilic în lichidele biologice prin metoda CGL.
2. Condiţiile de identificare a alcoolului în lichidele biologice prin CGL.3. Obiectele de cercetare asupra alcoolului etilic în analizele chimico-juridiciare
şi chimice.4. Cum se măsoară valoarea “timpului de reţinere” pe cromatogramă – de la
momentul introducerii probei, de la maximul picului de aer?5. Ce numim timp de reţinere corectat şi relativ?6. Ce detector se foloseşte la determinarea alcoolului etilic în sânge şi urină prin
metoda CGL?7. Împiedică oare determinării alcoolului etilic în lichidele biologice prin
metoda CGL alte componente “volatile”?8. Explicaţi construirea curbei de calibrare pentru determinarea etanolului în
sânge şi urină.9. Ce luăm în calitate de standard intern la determinarea alcoolului etilic?10.De ce se calculează coeficienţii de calibrare la determinarea etanolului în
sânge şi urină?
Răspundeţi argumentat la următoarele întrebări:1. Ce proprietăţi ale componentelor separate acţionează la timpul de reţinere în
baza staţionară.
37

2. Explicaţi de ce compuşii organici nesaturaţi posedă un timp de reţinere mai mare în faza staţionară polară decât în cea nepolară?
3. Cum are loc cromatografierea în faza staţionară lichidă benzenul şi fenolul? (TR)?
4. Explicaţi prioritatea metodei CGL faţă de celelalte metode.5. De ce depinde eficacitatea repartiţiei componentelor în coloană?6. Ce parametrii ai picului se folosesc în determinarea cantitativă?7. Ce se soluţionează efectuând analiza cantitativă prin metoda cromatografiei
gaz-lichide.8. Cum se construieşte graficul de calibrare prin metoda standardului intern,
calibrării absolute?9. De ce prin aplicarea metodei de dozare a standardului intern se întroduce în
cromatograf diferite volume de probă? 10.Cum determinaţi conţinutul etanolului în urină, dacă după analiza
cromatogramei înălţimea picului de etilnitrat hx=10 mm, înălţimea picului de propilnitrat 15 mm, R=1, Ku=1,03.
11.Determinaţi conţinutul etanolului în sânge, dacă înălţimea picului etilnitrat 25 mm, propilnitrat 17 mm, R=1, Ks=1,90.
12.Determinaţi Cu=Cs (după problema 1,2) şi trageţi concluzie de gradul de alcoolizare.
Lucrul practic al studenţilor la lucrarea de laborator Studenţii primesc cromatograme pentru rezolvarea 4 situaţii. Cromatogramele se înscriu în caiete. Pe una din ele se demonstrează ce parametri pe cromatograme se determină.
Studentul e dator să cunoască diferite moduri de calcul a suprafeţei picurilor:
B unde h – înălţimea picului S = h --------- B – baza triunghiului sub pic.
2
S = h unde h – înălţimea picului - lăţimea picului la jumătatea înălţimii
Studenţii îndeplinesc 4 însărcinări după cromatogramele expuse. Metoda normării ariilor
38

Fiecare student determină componenţa procentuală a amestecului (o cromatogramă) prin metoda normării ariilor de dozare a cromatogramelor. Pentru acesta se calculează suprafeţele tuturor picurilor.
Formula de calcul:SA
A = ----------- 100 S
Metoda standardului internCalculaţi conţinutul în % a unui component din amestecul medicamentos
prin metoda standardului intern (calculele se efectuează conform formulei):Sx Kx
A% = ----------- R 100Sst Kst
În cazul dat Sst şi Kst = 1, iar m standardului
R = ------------------------------------------------- m amestecului de analizat (fără standard)
Fiecare student calculează independent, reieşind din datele cromatogramei, formula având următorul aspect:
Sx
A% = ---- R 100Sst
Cromatograma se prelucrează calculativ. Toate datele se includ în tabelă.Nr croma-togra-mei
Com-ponent a ame-stec
Analiza cantitativă Analiza calitativăh S %
normei interne
Metoda standardulu
i
L Viteza lentei
TRTrel
Calcularea vitezei optime a gazului vector pentru repartiţia componenţelor.
Fiecare student primeşte cromatograme a substanţei de analizat la diferite viteze a gazului vector-heliu.a) pentru fiecare cromatogramă calculaţi numărul talerelor teoretice după
ecuaţia: x
N = 16 (----)2
Y
39

b) Calculaţi ÎETT după formula următoare : L cm
ÎETT= -------- N
unde ÎETT- înălţimea eficacităţii talerelor teoretice;L - lungimea coloanei cromatografice (arătată pe cromatogramă);N – numărul talerelor teoretice.
c) Construiţi graficul dependenţei ÎETT (axei ordinatelor) în raport cu viteza gazului vector (axei absciselor). Determinaţi viteza maximă a heliului după grafic, corespunzător ÎETT minime pe grafic.Alcătuiţi tabela.VHe X Y X
-----Y
X(----)2
Y
N ÎETT
Calcularea reproductibilităţii metodei CGL.Fiecare student primeşte 3 cromatograme a unii component, scrie condiţiile
experimentului şi măsoară înălţimea picului în mm, calculează înălţimea medie, greşala determinării separate, abaterea relativă medie.
Abaterea determinării separate:hmed – h1 = a1
hmed – h2 = a2
hmed – h3 = a3
Abaterea medie a1 + a2 + a3
--------------------- = B 3
Abaterea relativă medieB
----------- 100hmed
Abaterea medie relativă nu trebuia să depăşască 3-4 %.
Tema: Determinarea alcoolului etilic în sânge şi urină prin metoda CGL Prelucrarea metodică
40

Lucrul practic al studenţilor în laboratorCondiţiile de lucru: Aparat – Crom-4: coloană de oţel (sticlă) 2 m, faza
staţionară – PEG-1500, umplutura – cromaton cu argint N-AW-DMAS, T0coloanei - 680, T0 termostatului – 700, T catarometrului – 1000, intensitatea curentului în detector – 100 am, gazul vector – heliu 2,4 l/oră, sensibilitatea înscrierii 2, mărimea probei 2 ml. Cloralhidratul, formaldehida, acidul acetic, fenolul, dioxanul, acidul cianhidric, metilsalicilatul, metilmercaptofos, tiofos picuri nu formează.
Picurile metilnitrilului, hexanului, pentanului se înregistrează înaintea etilnitrilului, cloroformul, tetraclorura de carbon, dicloretanul, acetona, eterul, benzenul, toluenul, xilolul, octanul, propil-, izopropil-, butil-, izobutil, amil-, izoamilnitriţii se înregistrează după etilnitrit şi nu împedică identificării. Pentru a dovedi prezenţa alcoolului etilic ne folosim atît de timpul de reţinere corectat cît şi de timp de reţinere relativ.
Identificarea alcoolului etilic în amestecul model (scrieţi chimismul reacţiilor)În flacon de penicilină se introduc 0,5 ml sol acid tricloracetic 50%, 5 ml
sol n-alcoolpropilic (4 %) şi 0,5 ml sânge ori urină (ori C2H5OH), flaconul se închide cu un dop de cauciuc şi se fixează în cilindru metalic. Capacul cilindrului se închide în aşa mod ca dopul de cauciuc să închidă ermetic flaconul. Conţinutul flaconului se agită cu precauţie.
Un flacon se întroduce cu seringa de 1 ml, 0,25 ml soluţie nitrit de sodiu 30 %. Flaconul se agită şi peste 1 minută cu o altă seringă se colectează 2 ml probă gazoasă şi se introduce în injectorul cromatografului. Pe cromatograma înregistrată se determină timpul de reţinere corectat şi relativ. Timpul de reţinere relativ se determină după n-alcool propilic.
Determinarea cantitativă prin metoda standardului intern. Metoda grafică. Construirea curbei de calibrare a soluţiei standard de alcool etilic în lichidele biologice ori soluţie apoasă de etanol.
Fiecare student primeşte 4 cromatograme ale etilnitritului, corespunzător conţinutului de etanol în lichidele biologice (sânge ori urină). Fiecare cromatogramă se prelucrează după calculele de determinare calitativă şi cantitativă a alcoolului etilic în lichidele biologice. Pentru aceasta se calculează timpul de retenţie relativ a etilnitrilului. Pentru determinarea cantitativă se determină înălţimea picurilor de etilnitrit (hx) şi standard - n-propilnitrit (hst) şi se calculează raportul hx / hst 100. Se construieşte graficul – pe axa ordinatelor se înscrie raportul hx / hst 100, pe axa absciselor – concentraţia etanolului în lichidele biologice în promile (o/oo).
Soluţiile standarde ale etanolului în sânge şi urină nu sunt stabile şi pe ele în practica de lucru e nevoie să le renovăm. Mai corect ar fi să folosim soluţiile standarde apoase ale alcoolului etilic pentru construirea curbei de calibrare. Prin folosirea datelor acestui grafic se folosesc numaidecât coeficienţii de calibrare, care pentru determinarea etanolului în sânge este egal 0,95, în urină – 1,05.
41

Cs = C(H2O ) 0,95, Cur = C(H2O) 1,05 (CH2O – valoarea concentraţiei etanolului găsită după curba de calibrare a soluţiei apoase de etanol).
Metoda de calcul pentru determinarea cantitativă a etanolului în cazul aplicării standardului intern
Analiza cantitativă a etanolului după metoda standardului intern se efectuează şi prin metoda de calcul fără a construi graficul de calibrare, conform următoarei formule:
Sx
С‰ = ------- K R ; R = 1 Sst
K - coeficientul de corecţie relativ a suprafeţelor picurilor (Sx, Sst), determinat pentru fiecare component în raport cu standardul în condiţiile date. (FLS, temperatura de evaporare. Temperatura de termostatare, detectorul, viteza gazului, vector). De aici reiese calculul coeficientului de corecţie care se exprimă după formula: Sst
K = ------- С‰, ori suprafaţa lui se înlocueşte cu înalţimea picului. Sx
hst (înălţimea picului propilnitrit)K = ------------------------------------------ СC2H5OH hx (înălţimea picului etilnitrit)
Pentru calcularea K folosim soluţiile apoase de etanol (1‰, 2‰, 3‰, 4‰) cu standardul intern – alcoolul propilic. Soluţiile apoase a etanolului se prelucrează după metodică pentru dozare şi se cromatografiază. Pe cromatogramă se măsoară înălţimea picului etilnitrit şi propilnitrit (în soluţie apoasă de etanol) şi se calculează K, apoi se înmulţeşte la 0,95 ori 1,05. În rezultat coeficientul final, care va fi folosit în formula de calcul pentru sânge Ks
= Kmed 0,95, pentru urină Kur = Kmed 1,05.Exemplu de prelucrare a rezultatelor obţinute:
Conc. etanol Cx
Înălţimea
picului
etilnitrit
Înălţimea picului
propilnitrit
hst K = ------- Сx
hx Kmed
Ks
Kur
1‰2‰3‰4‰
6280140192
13599109106
135/62 1 = 2,1899/80 2 = 2,48109/104 3 =
2,34Kmed
0,95Kmed
1,05
42

5‰ 178 78 108/192 4 = 2,20
78/178 5 = 2,25
2,292,11 2,41
Exemplu de calcul:Problema 1 – Calculaţi cantitatea de etanol în sânge, dacă:hС2H5ONO = 60 mmhC3H7ONO = 130 mm 60
С‰ С2H5OH = ------ 2,11 = 0,97 % 130
Problema 2 – Calculaţi conţinutul etanolului în urină, dacă :hС2H5ONO = 175 mmhC3H7ONO = 80 mm 175
С‰ С2H5OH = ------ 2,41 = 5,27 % 80K – coeficientul de corecţie pentru fiecare concentraţieK- coeficientul mediu pentru determinarea conţinutului alcoolului etilic în sânge şi urină Controlul total3. Aprecierea rezultatelor obţinute în urma efectuării lucrului practic.4. Semnarea proceselor verbale. Bibliografie7. Materialul prelegerilor 8. Бабилев Ф.В., Тряпицына Т.П. Газожидкостная хроматография в
фармацевтическом анализе. Кишинев, “Штиинца”, 1978, р. 4-10, 15-19. 20-25. 30-53.
9. В.Ф. Крамаренко « Токсикологическая химия», Киев, Выща школа, 1989
10.Г.Мак. Нейр, Э.Бопелли. Введение в газожидкостную хроматографию. М., Мир, 1970. Р. 29-39.
11.Вигдергаиз М.С. Качественный газохроматографический анализ, гл. 3, М., “Химия”, 1979, р. 356.
12.Н.Перцев. Н. Корцев. Справочник по газовой хроматографии. Москва, “Мир”, 1987, р.246.
Lucrarea N 3Tema: Expertiza chimico- toxicologica a toxicilor volatili din materialul biologic. Determinarea alcoolului etilic în lichidele biologice prin metoda cromatografiei gaz-
43

lichide. “Metoda cromatografiei gaz-lichide în identificarea şi dozarea toxicilor “volatili”. Îndeplinirea buletinului de expertiză.
1. Scopul temei: Întărirea cunoştinţelor teoretice şi practice asupr analizei chimico- toxicologice a toxicilor volatili din materialul biologic. A studia metodica de dozare a etanolului în sânge şi urină prin aplicare metodei cromatografiei de gaze.
Determinarea gradului de alcoolizare în organism.
Scopuri practice:1. A deprinde efectuarea izolării toxicilor volatili din materialul biologic, aplicînd
metoda antrenării cu vapori de apă.2. A studia metodele de identificare a toxicilor volatili din distilatul obţinut.3. Determinarea cantitativă a toxicilor volatili.4. A studia biotransformarea toxicilor volatili. 5. A studia metodica de dozare a etanolului în sânge şi urină prin aplicare metodei
cromatografiei de gaze.6. Determinarea gradului de alcoolizare în organism.
Întrebările teoretice:1. Care este principiul metodei de izolare a toxicilor volatili?2. Explicaţi prepararea probelor preliminare în analiza chimico- toxicologica a toxicilor
volatili.3. Cum se izolează etilenglicolul şi acidul acetic din materialul biologic?4. Particularităţile izolării alcoolului izoamilic, etilic, metilic, fenolului, dicloretanului,
aldehidei formice.5. Analiza chimico- toxicologică a acidului cianhidric şi a sărurilor lui în extrasele
biologice.6. Însemnătatea toxicologică, mecanismul acţiunii toxice şi metabolismul acidului
cianhidric şi sărurilor lui.7. Analiza chimico- toxicologică, identificarea şi dozarea halogenderivaţilor(cloroform,
dicloretan, cloralhidrat, tetraclorura de carbon) în materialul biologic.8. Acţiunea toxicologică a derivaţilor halogenaţi ai hidrocarburilor.9. Caracteristica alcoolului metilic, etilic, izoamilic şi etilenglicolului. Particularităţile
izolării, identificării şi dozării alcoolilor în dependenţă de obiectul de cercetare. 10. Particularităţile acţiunii alcoolului etilic. Coma alcoolică.11. Metodele chimice de confirmare a prezenţei formaldehidei în organele cadaverice.12. Acţiunea toxică şi biotransformarea formaldehidei în organismul uman.13. Acetona şi acidul acetic. Toxicocinetica şi toxicodinamica.14. Analiza chimico- toxicologică a acidului acetic şi acetonei.15. Particularităţile cercetărilor chimico- toxicologice asupra fenolului şi derivaţilor săi.16. Biotransformarea fenolului şi acţiunea toxică a lui.17. Expertiza intoxicaţiilor alcoolice în materialul cadaveric. Factorii ce influenţează
asupra determinării etanolului.18. Tetraetilul de Pb şi particularităţile cercetării produselor petroliere asupra acestui
toxic. Mecanismul acţiunii toxice.19. Analiza ch.- toxicologică a nitrobenzenului, anilinei.20. Mecanismul acţiunii toxice a nitroderivaţilor.21. Absorbţia, repartiţia şi eliminarea cianurilor şi acidului cianhidric, cloroformului,
cloralhidratului, tetraclorurii de carbon, dicloretanului, formaldehidei, acetonei, ac. acetic, alcoolilor alifatici, etilenglicolului, fenolului, nitrobenzenului.
44

22. Biotransformarea ac. cianhidric, derivaţilor halogenaţi, ac. acetic, formaldehidei, etilenglicolului, hidrocarburilor saturate.
23. Absorbţia, distribuţia şi eliminarea alcoolului etilic în organismul uman.24. Biotransformarea alcoolului etilic.25. Simptomatologia intoxicaţiilor cu alcool etilic.26. Expertiza intoxicaţiilor cu alcool etilic.
a) Interpretarea rezultatelor expertizei în dependenţă de materialul biologic.27. Intoxicaţiile mixte de alcool etilic.28. Caracteristica metodei gaz-cromatografice în vederea determinării toxicilor “volatili”.
Părţile componente ale cromatografului.29. Tipurile de detectori. Principii de lucru.30. Explicaţi eficacitatea coloanei.31. De ce depinde selectivitatea fazei staţionare?32. Daţi definiţia timpului corectat şi timpului relativ în coloană.33. Principiu de identificare a toxicilor volatili prin metoda cromatografiei de gaze.34. Dozarea toxicilor volatili prin metoda cromatografiei de gaze. Principiu de lucru şi
metodele de calcul.35. Dozarea etanolului în lichidele biologice – chimismul, metodica, calculele.
Îndeplinirea scopului practicCondiţiile experimentului: aparatul “Crom-4”, coloana de sticlă – 2 m, faza staţionară
PEG-1500 10 %, T0 colanei – 600C, T0 camerei de evaporare 700 C, T0 catarometrului – 1000C, intensitatea curentului – 100 am., gazul purtător – Heliu, viteza 2,4 l/oră, mărimea probei – 2 ml.
Problema 1Determinarea coeficientului de corecţie pentru analiza cromatografică a lichidelor
biologice la prezenţa alcoolului etilic prin metoda de calcul.Fiecare student primeşte soluţie în apă (cu conc. 1-5 ‰) şi prelucrează soluţia după
metodica [3], după aceea cromatografiază amestecul de gaze în aparatul “Crom-4”. Pe cromatograma obţinută se măsoară înălţimea picului etilnitrit şi propilnitrit. După datele obţinute se efectuează calculele coeficientului de corecţie relativ după formula:
hst (înălţimea picului propilnitrit)1) K = ------------------------------------------ СC2H5OH ‰
hx (înălţimea picului etilnitrit)
2) Se calculează Kmed
3) Se calculează coeficientul de corecţie pentru determinarea etanolului în sânge în condiţiile cromatografice date luând în vedere coeficientul de calibrare.
K sânge = Kmed · 0,954) Se calculează coeficientul de corecţie pentru determinarea etanolului în urină în
condiţiile experimentului, luând în vedere coeficientul de calibrare.K ur = Kmed · 1,05
Problema 2Dozarea alcoolului etilic în sânge şi urină prin metoda de calcul.
Fiecare 2 studenţi primesc problemă – două flacoane cu lichid biologic – urină şi sânge cu conţinut de alcool etilic.
Probele se prelucrează pentru dozarea alcoolului etilic (Problema 1) şi se cromatografiază produsele gazoase ale reacţiei în aparatul “Crom-4”. Cromatograma obţinută se prelucrează calitativ (distanţa de reţinere L); TR – corectat, Trel – timpul relativ. Se
45

efectuează dozarea alcoolului etilic prin metoda de calcul, pentru care se măsoară înălţimea etilnitritului şi propilnitritului şi se efectuează calculele.
Înălţimea picului propilnitritС etanolului(sînge) ‰= --------------------------------------- Ksânge
Înălţimea picului etilnitrit
Înălţimea picului propilnitritС etanolului(urină) ‰= --------------------------------------- Kurină
Înălţimea picului etilnitrit
Problema 3Determinarea etanolului prin metoda standardului intern după graficul de calibrare.
a) Pentru îndeplinirea acestei probleme studenţii folosesc datele din I problemă, îndeplinesc tabela, după datele obţinute construiesc graficul de calibrare.
Conc. etanolului
în ‰
Înălţimea picului etilnitrit
h -C2H5ONO
Înălţimea picului propilnitrit
h –C3H7ONO
h C2H5ONO ----------------- 100
h –C3H7ONO1‰2‰3‰4‰
b) Determinarea etanolului în lichidele biologice după graficul de calibrare. Se prelucrează sângele şi urina după metodica soluţiei apoase de etanol. Se măsoară pe cromatogramă
h -C2H5ONO şi h –C3H7ONO şi se găseşte pe grafic concentraţia corespunzătoare a etanolului (CH2O). Deoarece graficul de calibrare e cunoscut după soluţia apoasă de etanol, concentraţia adevărată a etanolului în sânge Csânge = CH2O 0,95
în urină Curina = CH2O 1,05În sfârşit se compară datele obţinute după graficul de calibrare şi metoda de calcul.Se fac concluzii despre conţinutul etanolului în sânge şi urină şi gradul de alcoolizare.
Bibliografie1. Conspectul lecţiilor2. T.Brodicico, V.Valica “Curs de Chimie toxicologică”, Chişinău, 20033. Крамаренко В.Г. Токсикологическая химия. – Киев: Выща школа, 1989, с.135-143.4. Лужников Е.А. Клиническая токсикология. М. «Медицина», 1994, с. 5525. Белова А.В. Руководство к практическим занятиям по токсикологической химии.
Москва: «Медицина», 1976, с. 37-44
Buletin de expertiză Actul analizei chimico- toxicologice a corpurilor delicte, conform dosarului cetăţeanului X, transmise anchetatorului__________
46

Analiza se efectuează în laboratorul toxicologic al Universităţii de Stat de Medicină şi Farmacie ”Nicolae Testemiţanu” de chimistul- expert__________
Începutul___________ Sfîrşitul__________
Examenul exteriorPentru cercetare se ia un borcan de sticlă albă, cu volumul de 500 ml, acoperit cu permanganat, cu inscripţia „Borcanul Nr. 1- bucăţi de ficat şi rinichi”. Conţinutul probei prezintă bucăţi de organe indicate cu masa: ficat- 250 g, rinichi- 200 g. Reacţia conţinutului probei (turnesol- neutră). La miros- nimic deosebit.
Cercetatrea chimică100 g material biologic din borcanul Nr. 1 se amestecă cu apă distilată pînă la o consistenţă densă, se acidulează cu acid oxalic pînă la pH slab acid (hîrtia indicator turnesol) şi se supune antrenării cu vapori de apă: 1- a fracţie a distilatului în volum de 3 ml s- a adunat într- un volum de 5 ml soluţie NaOH1%. Fracţia a 2- a (25 ml) şi a 3- a (25 ml) se adună separat fără adaos de NaOH.
Cercetarea distilatului1. La 1- a fracţie de distilat se adaugă cîteva picături de sulfat de Fe şi 1- 2 picături sol.
clorură de Fe, apoi lichidul se acidulează cu HCl pînă la pH slab acid. Nici deodată, nici peste 48 ore precipitat albastru n- a apărut.
2. La a 2- a fracţie se face proba cu sol. de rezorcină în NaOH, care se încălzeşte, dar nu se observă coloraţie.
3. La a 2- a fracţie se efectuează proba cu sol. de Iod în NaOH. La încălzire pe baia de apă cade precipitat galben cu miros de cloroform.
4. La o parte din fracţia a 2- a se adaugă volum egal de sol. ac. sulfuric 20% şi cîteva cristale KMnO4. Peste un interval de timp se simte miros plăcut de fructe. Peste 20 min., lichidul se filtrează şi cu filtratul incolor se efectuează reacţiile următoare:
a.la jumătate din filtrat se adaugă 1/5 volum ac. sulfuric conc., în care au fost solubilizate cîteva cristale de morfină. Culoare albastră- violetă nu s- a observat. b.la a 2- a jumătate de volum a filtratului se adaugă acelaşi volum de ac. fuxin sulfuros şi 1 ml ac. sulfuric conc.. Nu se observă nici o coloraţie.5. Cîteva cristale de acetat de Na se dizolvă la 1 ml sol. din distilatul 2, care se
amestecă cu un volum dublu de ac. sulfuric conc. La o uşoară încălzire se simte miros de etilacetat.
6. O parte din distilatul 2 se amestecă cu acelaşi volum sol. alcoolică 5% NaOH şi se încălzeşte atent timp îndelungat, la răcire se adaugă ac. azotic şi apoi se adaugă sol. nitrat de Ag 5%. Formarea opalescenţei şi precipitatului nu se observă. Reziduurile distilatului 2 se amestecă cu al 3- lea şi se supun cercetărilor.
7. La o parte din distilat se adaugă cîteva picături apă de Br, unde imediat se formează opalescenţă gălbuie; o jumătate din distilat se supune deflegmaţiei de 2 ori. După a 2- a deflegmaţie s- a colectat în 1- l distilat 5 ml lichid, în al 2- lea 10 ml lichid. Cu 1- l distilat s- au efectuat reacţiile descrise în p. 3, 4, 5 cu acelaşi rezultate, la efectuarea reacţiei descrise în p. 5 mirosul de etilacetat devine mai pronunţat. În urma reacţiei cu apa de Br, opalescenţa nu se observă.
8. A 2- a parte a mestecului distilat se unesc, se alcalinizează cu carbonat de Na şi se repetă extracţia cu eter. Extracţiile eterice se unesc, se filtrează, iar eterul se înlătură la
47

temperatura camerei. Reziduul a fost neînsemnat. După solubilizarea lui în 2- 3 picături apă distilată se adaugă 2 picături clorură de Fe, proaspăt pregătită. Culoarea violetă nu se observă.
Concluzie Din analiza efectuată şi descrisă rezultă că în materialul biologic al cetăţeanului X s- a
identificat alcool etilic. Nu s- au identificat: ac. cianhidric, alcoolul metilic, cloroformul, tetraclorura de
carbon, cloralhidratul, formaldehida, fenolul, alcoolul izoamilic.
.
48


















