
LUCRARE ȘTIIȚIFICĂ: REVIEW -...
18
LUCRARE ȘTIIȚIFICĂ: REVIEW “Terapia cu hormoni de creștere în sindromul Prader-Willi” Autori: Antonia Gherlan 1 Cristiana Voicu 1 Co-autor: Dr. Ursu Radu Ioan 2 , medic specialist Genetică Medicală Coordonator: Mihaela Stancu 3 , medic rezident Psihiatrie Pediatrică Afilieri: 1. Universitatea de Medicină și Farmacie “Carol Davila”, Facultatea de Medicină Generală, MG an II 2. Catedra de Genetică Medicală, UMF ”Carol Davila”, București 3. Spitalul Clinic de Psihiatrie “Prof. Dr. Alexandru Obregia”, UMF “Carol Davila”, București
Transcript of LUCRARE ȘTIIȚIFICĂ: REVIEW -...

LUCRARE ȘTIIȚIFICĂ: REVIEW
“Terapia cu hormoni de creștere în sindromul Prader-Willi”
Autori: Antonia Gherlan1
Cristiana Voicu1
Co-autor: Dr. Ursu Radu Ioan2, medic specialist Genetică Medicală
Coordonator: Mihaela Stancu3, medic rezident Psihiatrie Pediatrică
Afilieri:
1. Universitatea de Medicină și Farmacie “Carol Davila”, Facultatea de Medicină Generală, MG
an II
2. Catedra de Genetică Medicală, UMF ”Carol Davila”, București
3. Spitalul Clinic de Psihiatrie “Prof. Dr. Alexandru Obregia”, UMF “Carol Davila”, București

2
CUPRINS
I.INTRODUCERE…………………………………………………….……………………...4
I. A. GENERALITĂȚI……….............................................................................................................. 4
I.A.1. Premise………………………………………………………………….. ............................ 4
I.A.2. Cauzele………………………………………………………………….. ............................ 4
I.A.3. Semnele și simptomele………………………………………………….. ............................ 4
I.A.4. Co-existența sindromului Prader-Willi și Angelman………………... ................................ 5
I. B. DIAGNOSTIC .............................................................................................................................. 6
I.B.1. Diagnostic clinic .................................................................................................................... 6
I.B.2. Diagnostic genetic ................................................................................................................. 7
I. C. MOTIVE ..................................................................................................................................... 7
I. D. SCOP .......................................................................................................................................... 8
II. PARTE PRINCIPALĂ…………………………………………………………...……….8
II. A. MANIFESTĂRI ENDOCRINE; CORELAȚII PRIVIND TERAPIA CU STH……… ................. 8
II.A.1. Hipogonadismul ................................................................................................................... 8
II.A.2. Insuficiența glandelor suprarenale……………………………………... ............................ 8
II.A.3.Hipotiroidismul…………………………………………………………... .......................... 8
II.A.4 .Metabolismul glucozei și diabetul………………………………………. .......................... 9
II.B. TERAPIA CU HORMONI DE CREȘTERE……………………………………… ................... 10
II.B.1. Generalități ......................................................................................................................... 10
II.B.2. Hormoni de creștere……………………………………... .................................... 10
II.B.3.Tratamentul hormonal în funcție de vârsta de debut…………………………… .. 11
II.B.3.1. Tratamentul hormonal la copii……………………………..……………...11
II.B.3.2. Tratamentul hormonal la adulți…………………………….………….......11
III. DISCUȚII ȘI CONCLUZII……………………………………………………….……13
III.A. DISCUȚII ................................................................................................................................. 13
III.A.1.Amprentarea genomică și disomia uniparentală………………………….…….. ............. 13
III.A.2.Comportamentul obsesiv-compulsiv………………………….…….. .............................. 14
III.A.3. Durata tratamentului ......................................................................................................... 14
III.A.4.Prognostic și rata de mortalitate……………………………………. ................................ 14
III. B. CONCLUZII ........................................................................................................................... 15
IV. BIBLIOGRAFIE………………………………………………………………….…….16

3
ABSTRACT
Sindromul Prader Willi este o boală genetică complexă cauzată de o deleție de la nivelul
cromozomului 15 patern, brat q (15q11.2-q), cu o incidență de 1 la 12.000/1 la 15.000 dintre
nou-născuții vii. Tabloul clinic este unul vast, incluzând disfuncții metabolice și endocrine cu
obezitate progresivă, retard mintal, statură mica, lipsa centrului sațietății, hipotonie musculară și
tulburari comportamentale. Totodată, acest sindrom a fost diagnosticat la pacienți de orice rasă,
sex, cu vârste de debut variabile.
Acest review își propune realizarea unei analize obiective a evoluției bolii în urma
terapiei cu hormoni de creștere și efectele acesteia pe termen scurt și lung, trecând în revistă
avantajele, respectiv dezavantajele, survenite în urma administrării tratamentului.
Dintre beneficiile constatate în urma terapiei putem aminti: creșterea în înalțime și a
masei musculare cu scăderea procentului de țesut adipos corporal, îmbunătățire cognitiv-
emoțională. Pe termen lung, totuși, au fost semnalate cateva puncte slabe: tratamentul cu
hormoni nu poate compensa suficient deficitul de masă musculară, nu scade apetitul, poate
agrava patologii preexistente, ca de exemplu probleme respiratorii de origine multifactorială cu
complicații de tip insuficiență respiratorie sau disfagie. Totodată, începerea și încheierea
tratamentului nu pot fi stabilite cu precizie, fiecare caz fiind unic și având un răspuns diferit.
În concluzie, terapia cu hormoni de creștere este una dintre singurele metode disponibile
la ora actuală cu potențial benefic mare, deși are efecte limitate și în anumite cazuri poate agrava
starea pacientului.
CUVINTE CHEIE: Sindrom Prader-Willi (SPW), sindrom Angelman (SA), hormon de creștere
uman (hGH), obezitate, tratament, hiperfagie, hipotonie, insuficiență hipotalamo-hipofizară.

4
I.INTRODUCERE
I. A. GENERALITĂȚI
A.1. Premise: Sindromul Prader Willi este o boală genetică complexă, cu o incidență de
1 din 12.000 până la 1 din 15.000 nou-născuți vii, care afectează ambele sexe, toate rasele și are
un debut diferit, mulți dintre pacienți nefiind diagnosticați la timp, alții niciodată. Această boală
este cea mai reprezentativă formă de obezitate sindromatică, pacienții având disfuncții endocrine
și metabolice, simptomul definitoriu fiind hiperfagia datorată absenței centrului hipotalamic al
sațietății[1]
.
A.2. Cauzele apariției acestei boli sunt datorate unor erori structurale de la nivelul
cromozomului 15 patern, acestea având ca natură fie absența unor gene de pe bratul q (aberații
cromozomiale de tip microdeleții), aproximativ 70-80%, fie prezența genelor amprentate
(imprinting genes). Alteori, se poate ca pacientul să fi moștenit două copii ale cromozomului 15
matern și să lipsească cel patern - disomie uniparentală, în 10-15% din cazuri.
A.3. Semnele și simptomele apar diferit în funcție de etapele creșterii pacientului, în
perioada neonatală și de sugar fiind caracteristice următoarele: tonus muscular slab, facies
caracteristic (ochi migdalați, buză superioară subțire, îngustarea diametrului bifontal), hipoplazia
organelor genitale, supt dificil, întârzieri de limbaj și de dezvoltare motorie generală. În perioada
copilăriei până la viața de adult se vor remarca: creșterea apetitului (hiperfagie), deficit statural,
tulburări de somn, retard mintal moderat și afectare psihocomportamentală cu crize de isterie,
tendințe obsesiv-compulsive, frecvent și dermatilomanie – lezarea voluntară a tegumentului. Pot
interveni și alte probleme endocrine: hipotiroidism, deficit de hormoni de creștere, insuficiență
adrenală. Majoritatea manifestărilor clinice pot fi cauzate de disfuncția hipotalamică, incluzând
hiperfagia, temperatura instabilă, prag de durere ridicat și probleme de respirație în somn.
Disfuncția hipotalamică dă naștere adesea și unei variabile insuficiențe hormonale ale
hipofizei.[2]
Așadar, boala prezintă un tablou clinic complex, cu variate complicații, care pe parcursul
vieții vor necesita o constantă colaborare cu cadrele medicale pentru a stabili tratamente adecvate
și a-i oferi pacientului o viață cât mai bună. Din acest motiv, screening-ul are un rol-cheie în
ameliorarea unor simptome, iar în cazul pacienților cu Prader Willi, obezitatea progresivă este
principală problemă a acestora, care trebuie ținută sub o atentă observație deoarece are tendințe
de morbiditate.
A.4. Co-existența sindromului Prader-Willi și Angelman
Sindromul Prader-Willi și sindromul Angelman (SA) reprezintă două maladii distincte
din punct de vedere clinic, asociate cu multiple anomalii și cu retard mintal, însă care sunt

5
discutate împreună deoarece împart o cauză similară și mai puțin frecventă: acestea antrenează
gene localizate în aceeași regiune în genom, mapate cromozomului 15 (q11-q13).[3]
Tulburările
endocrine de comportament sunt mult mai severe în sindromul Prader-Willi decât în SA.
Figura 1: Rezumat al hărții si expresiei genetice a regiunii cromosomale 15q11.2-q13 (după
4)
Regiunea cromozomială a sindromului Prader-Willi (marcată cu albastru) conține 5 copii unice ale
expresiei genelor (regiunea PWS) care codifică pentru polipeptidele (MKRN3, MAGEL2, NECDIN și SNURF-
SNRPN) și o familie compusă din 6 gene paterne ARNsno. Doar UBE3A și ATP10A (marcate cu portocaliu), care
au legătură cu sindromul Angelman au expresii specific materne. Centrul întipărit (IC) are o structură bipartită cu
o componentă AS (maternă, colorată în orange), respectiv una PWS (paternă, colorată în albastru). Centrul bipartit
se dispune în cadrul regiunii întipărite PWS/AS pe o distanță de 2.5-Mb. Grupul receptorilor genelor GABA
(GABRB3, GABRA5 și GABRG3), OCA2 (tipul II de albinism) și HERC2 nu sunt întipărite și conțin o expresie de
tip biparental (afișat în verde). Liniile verticale zimțate arată cele 3 puncte comune de deleție de 5-6 Mb ale PWS și
AS: BP1, BP2 și BP3. Mai rar întâlnite sunt punctele de rupere distale la BP4 și BP5. Între BP1 și BP2 se găsesc 4
gene adiționale neîntipărite NIPA1, NIPA2, CYFIP1 și GCP5. Delețiile de tip 1 (T1D) sunt extinse de la BP1 la
BP3, iar delețiile de tip 2 (T2D) se întind de la BP2 la BP3.
Ambele boli genetice pot rezulta din microdeleții, disomii uniparentale sau dintr-un
defect al centrului de întipărire în 15q11-q13, însă anormalitatea derivă din cromozomul 15
paternal pentru Prader-Willi, respectiv cromozomul 15 maternal pentru Angelman din cauza
amprentării genomice. Relația genetică dintre Sindromul Prader-Willi, respectiv sindromul
Angelman, le transformă în boli unice și cu potențial instructiv care contribuie substanțial la
populația care suferă de tulburări cognitive.[5]

6
I. B. DIAGNOSTIC
B.1. Diagnostic clinic
Diagnosticul postnatal are la bază tabloul clinic caracteristic, putând fi apoi confirmat de
testele genetice. Obezitatea morbidă infantilă este unul dintre semnele care alarmează pacienții.
Statura mică devine evidentă în copilărie și adolescență datorită deficitului de hormoni de
creștere, înălțimea medie ajungând la aproximativ 148 cm la fete și 150 cm la băieți[6]
.
Nutriția copilului, deși este în strânsă legătură cu obezitatea, nu este o cauză propriu-zisă,
ci o altă complicație a acestui sindrom, foarte greu de controlat și cu efecte psihologice pe
termen lung.
Tabel 1: Nutriția în dezvoltarea pacienților cu sindrom Prader Willi (după
[7])
Există anumite boli care aparțin fenotipului sindromului Prader Willi, de aceea un
element de bază este diagnosticul diferențial. Unul dintre exemple este hipotonia infantilă, care
poate fi congenitală, în cadrul atrofiei musculare spinală sau în sindromului Angelman etc.
Retardul mintal este o altă manifestare pe care o regăsim și in sindromul X fragil, în
Cohen și alte boli genetice, având un grad de severitate variabil.
B.2. Diagnostic genetic
Diagnosticul clinic este confirmat de investigațiile genetice: studii de metilare a ADN-
ului MS-PCR (methylation specific polymerase chain reaction), RT-PCR (real time polymerase
chain reaction) și testul FISH (fluorescence in situ hybridization).
a) citogenetic
Testul FISH este recomandat pentru vizualizarea delețiilor (cea mai frecventă cauză) și de
aceea este, de regulă, prima investigație în caz de suspiciune de sindrom Prader Willi.
Deși aceste analize sunt concludente, ele nu pot determina cu precizie dimensiunile
delețiilor, așadar nu se poate afla câte gene sunt implicate în erorile de la nivelul cromozomului
15.
b) molecular
Complementar cu acesta se poate realiza MS-PCR care va confirma boala cauzată de
Faza Vârsta Caracteristici clinice
0 Prenatal - naștere Activitate motorie scăzută și greutate mică la naștere
1a 0-9 luni Hipotonie și dificultăți în hrănire, apetit slab
1b 9-25 luni Hrănire mai bună și apetit
2a 2-4 ani Creștere în greutate fără ca apetitul sau caloriile să fie în exces
2b 4-8 ani Apetit crescut
3 8 ani - adult Hiperfagie, rareori apare senzația de sațietate
4 Viața de adult Lipsa senzației de sațietate

7
disomii uniparentale sau amprentare eronată a genelor.
Constatând limitarea tehnicii FISH, s-a încercat observarea într-un studiu a dimensiunilor
delețiilor prin metoda RT-PCR folosind probe de ADN izolat din leucocite periferice și din
celule epiteliale bucale, iar rezultatele au fost comparate cu cele ale investigațiilor standard pe
pacienți cu Prader Willi, concluzia studiului fiind ca metoda RT-CPR este eficientă[8]
.
I.C. MOTIVE
Sindromul Prader-Willi este o afecțiune genetică complexă, care însă sub atentă
supraveghere medicală și multidisciplinară poate avea un pronostic favorabil și o speranță de
viață ridicată. Necesitatea diferitelor terapii pentru prevenirea sau ameliorarea simptomatologiei
specifice acestei afecțiuni reprezintă unul dintre motivele abordării acestei teme.
Hipotonia musculară și creșterea deficitară în înălțime sunt 2 dintre elementele principale
prezente în tabloul clinic al pacienților cu SPW, iar una dintre puținele terapii benefice care pot
ameliora aceste deficiențe este reprezentată cu hormonul de creștere.
Motivul pentru care am ales să abordăm acest subiect a fost faptul că modul de viață al
acestor pacienți este unul precar, cu cauza principală obezitatea, din care derivă marea majoritate
a complicațiilor. În urma cercetările proprii estimăm faptul că în România există peste 50 de
pacienți cu această maladie genetică. Existența unui astfel de review se poate dovedi deci
benefică deoarece fiind o boală rară orice nouă cercetare pe această temă reprezintă o speranță
pentru acești pacienți.
I. D. SCOP
Obiectivul acestei lucrări este de a da o altă perspectivă atât a speranței de viață, cât și a
calității acesteia pentru pacienții care suferă de sindromul Prader-Willi. Tema propusă este
îndelung dezbătută la nivel mondial în sistemul medical, iar articolele la care vom face referire
pe parcursul prezentării vor fi selecționate din rândul celor care prezintă studii concrete și
omologate asupra atât a efectelor pozitive ale tratamentului în cauză cât și a efectelor negative,
dând la final o imagine de ansamblu a eficienței acestei terapii. Considerăm că selectarea unor
articole și studii de actualitate este necesară pentru formarea unui tablou obiectiv și o bună
înțelegere a efectelor tratamentului în cauză. Sursele principale de literatură au fost: PubMed,
Omim, Research Gate, fiind printre cele mai populare baze de date, la curent cu cercetările
făcute la nivel mondial.
Totodată, menționăm că această lucrare este efectuată în cadrul Programului Operational
Sectorial pentru Dezvoltarea Resurselor Umane (POSDRU), finantat din Fondul Social
European si Guvernul Romaniei prin contractul nr. POSDRU/156/1.2/G/141745.

8
II. PARTE PRINCIPALĂ
II. A. MANIFESTĂRI ENDOCRINE; CORELAȚII PRIVIND TERAPIA CU STH
A.1. Hipogonadismul este prezent atât la bărbați cât și la femei și se manifestă prin
hipoplazie genitală, dezvoltare incompletă la pubertate și, foarte important, infertilitate.
Hipogonadismul este de obicei asociat cu concentrații serice scăzute de gonadotropine și
cauzează o dezvoltare pubertală incompletă, întârziată și câteodată deficientă. Deși
hipogonadismul în sindromul Prader-Willi s-a considerat a fi în totalitate de origine
hipotalamică, constatări recente au sugerat o combinație între deficiențe hipotalamice și primare
gonadale , concluzie bazată în mare parte pe absența hipogonadotropismului și pe un nivel
anormal de inhibină B la pacienți afectați de ambele sexe.[9]
A.2. Insuficiența glandelor suprarenale
Insuficiența medulosuprarenalei este întâlnită in sindromul Prader-Willi, dar frecvența
este neclară. Copiii și adulții cu sindromul Prader-Willi prezintă un risc de insuficiență
suprarenală datorită disfuncției hipotalamice generalizate.
Într-o serie de cazuri de deces neprevăzut, autopsiile au relevat la 3 din 4 copii cu Prader-
Willi care prezentau febră sau alte boli acute, glande adrenale de dimensiuni mici având drept
criteriu greutatea.[2]
A.3. Hipotiroidismul
Insuficiența secreției glandei tiroide a fost raportată la aproximativ 20-30% dintre copiii
cu sindromul Prader-Willi. Un studiu recent la copiii cu PWS sub vârsta de 2 ani au arătat ca
72,2% prezintă anormalități pe axa hipotalamo-hipofizo-tiroidiană evidențiată printr-o
concentrație scăzută de tiroxină liberă sau totală în prezența unei valori normale de hormon
tireostimulator (TSH). De asemenea, un studiu al funcției tiroidiene realizat pe 75 de copii cu
PWS, care se aflau sub tratament cu hGH cu un dozaj de 1mg/m2/zi pe o perioadă de un an de
zile, a relevat o scădere semnificativă a nivelului de tiroxină liberă, în timp ce nivelul de
triiodotironină a rămas neschimbat. Este recomandat ca nivelurile de tiroxină liberă si TSH să fie
monitorizate în primele 3 luni de viață și după aceea anual, în special dacă pacientul primește
tratament cu hormon de creștere uman (STH).[2]
A.4 .Metabolismul glucozei și diabetul
Diabetul de tip 2 a fost raportat la 25% dintre adulții cu Prader-Willi cu un vârf la o
vârstă medie de 20 de ani. Diabetul și intoleranța la glucoză sunt mult mai puțin frecvente la
copiii cu PWS. Mai multe studii au demonstrat ca subiecții cu PWS care nu urmează terapie cu

9
hormoni de creștere prezintă un nivel scăzut de insulină și o sensibilitate mare la insulină având
drept cauză niveluri crescute de grelină pentru gradul de obezitate asociat și concentrații scăzute
de STH. De altfel, evaluarea riscului de obezitate este recomandat înaintea inițierii terapiei cu
hormoni de creștere la pacienții cu obezitate cu o vârstă peste 12 ani, urmând o supraveghere
periodică pentru cei care sunt sub tratament cu hGH.[2]
Într-un studiu realizat în România pe 5 pacienți suspectați de obezitate genetică (4 de sex
feminin, 1 de sex masculin, cu vârste între 5 și 19 ani) au fost urmăriți diferiți parametri: TSH,
GH, IGF-1, LH, FSH, corizol, hormoni sexuali, trigliceride, colesterol total, HDL etc. În ceea ce
privește rezultatele, la un pacient s-a constatat un ușor hipotiroidism (mai multe studii au arătat o
frecvență de hipotiroidism în Prader Willi de până la 25%), iar cortizolul bazal a fost în toate
cazurile în limite normale. S-ar părea că insuficiența adrenală are o incidență scăzută, totuși toate
aceste funcții trebuie monitorizate în permanență pentru a evita complicații ulterioare[10]
.
Criptorchidismul este o complicație care apare la 80-90% dintre pacienții de sex masculin
și este o tulburare în procesul de coborâre a testiculelor în scrot. Singurului pacient din studiu de
sex masculin i-a fost aplicat tratamentul chirurgical de la vârsta de 2 ani pentru a înlătura acestă
problemă. Deși pacienții din studiu aveau un nivel de hormoni de creștere la limita inferioară a
normalului, doi dintre aceștia, care nu prezentau contraindicații, au primit tratament hormonal (o
doză de 0.05 mg/kg/zi) care îmbinat cu un mediu înconjurător adecvat a dat rezultate bune:
dezvoltare psiho-motorie îmbunătățită, nu au avut efecte adverse.
II.B. TERAPIA CU HORMONI DE CREȘTERE
B.1. Generalități
În contextul unei boli cu un tablou clinic endocrin foarte complex, a fost recunoscută și
aprobată ca tratament recomandat în sindromul Prader Willi terapia cu hormoni de creștere
umani (hGH) pentru a restabili echilibrul acestei clase de compuși importanți în organism, cât și
pentru a îmbunătăți diferite efectele negative ale bolii: creșterea în înălțime, dezvoltarea masei
musculare, aspecte cognitiv-comportamentale și altele.
Tratamentul constă în administrarea unei doze zilnice de hormoni, calculată în funcție de
greutatea pacientului, teoretic pe o perioadă lungă de timp, sub atenta observație a clinicianului.
Deși primele studii s-au făcut începând cu anii 1990, în lumea medicală încă există
controverse în ceea ce privește vârsta recomandată de începere și de încheiere a tratamentului.
Vârsta optimă de debut nu este cunoscută, însă consesnsul experților este de a începe terapia
înainte de a se instala obezitatea, care de obicei are loc la vârsta de 2 ani. Unii experți recomandă
tratamentul încă de la primele 3 luni de viață.[11]
Așadar, rămâne la latitudinea endocrinologului

10
să stabilească dacă și când un pacient îl va începe. Aceste limitări, împreună cu contraindicațiile
care fac ca unii pacienți să nu fie eligibili unui asemenea tratament, sunt o parte din dezavantaje.
Orientările clinice recomandă o doză de început de 0.5 mg/m2/zi cu o creștere progresivă
a dozei până la 1 mg/m2/zi.
[11-13]
Terapia cu hGH nu este fără risc. Există contraindicații și posibile reacții adverse la
tratamentul cu hGH, acestea includ o categorie de maniestări care trebuie semnalate urgent:
dureri musculare, articulare sau nervoase, edem, sindrom de tunel carpian, amorțeli la nivelul
membrelor (parestezie) sau mâncărimi, niveluri crescute de colesterol. Totodată, această terapie
crește riscul apariției diabetului zaharat, dar și a proliferării celulelor canceroase, de aceea
pacienții care se prezintă cu una dintre aceste probleme nu vor fi eligibili pentru tratamentului cu
hGH.
B.2. Hormoni de creștere
Prevalența raportată cu privire la deficiența de hormoni de creștere în sindromul Prader-
Willi variază de la 40% la 100% în funcție de criteriul de diagnostic folosit, iar majoritatea
studiilor raportează o predominare către valoarea ridicată a procentajului.[14,15]
Manifestările
clinice asociate cu insuficiența de hormoni de creștere includ statura mică în ciuda obezității,
structură corporală anormală, niveluri scăzute de factor 1 de creștere insulinic (IGF-1) și secreție
scăzută de hormoni de creștere ca răspuns la testul provocat.[13]
În Europa, ”îmbunătățirea
structurii corporale” este inclusă în indicația aprobată pentru terapia cu hGH în sindromul
Prader-Willi. În practică, hGH este folosită în principal pentru beneficiile ei, altele decât
greutatea corporală crescută, incluzând îmbunătățirea structurii corpului și funcția motorie.[16]
B.3. Tratament hormonal în funcție de vârsta de debut
B.3.1. Tratamentul hormonal la copii
Un studiu (Eiholzer et al, 2004, Elveția) [17] efectuat în 2004 pe un grup de 11 copii cu
vârste mai mici de 2 ani a urmărit evoluția acestora înainte și în timpul terapiei cu hGH (timp de
30 luni) prin comparație cu un alt grup de 6 copii cărora le-a fost administrată coenzima Q(10)
timp de 1 an. Concluziile au fost favorabile terapiei, constatându-se o ameliorare în acumularea
de țesut adipos și, deci, un posibil control al obezității.
Pe un grup de 25 de copii cu vârste între 4 și 37 de luni s-a realizat un alt studiu (Myers
SE et al., 2007, SUA) [18]. S-au ales aleatoriu 2 grupuri: primului i s-a administrat hGH timp de 2
ani (1 mg/m(2)/zi), iar celui de-al doilea i s-a administrat tratamentul după 1 an (1.5 mg/m(2)/zi).
Rezultatele au fost următoarele: pe subiecții din primul grup, prin comparație cu al doilea grup,
s-au constatat ameliorări în depunerea de țesut adipos, o creștere mai mare a capului, vorbire
îmbunătățită. Tratamentul a fost tolerat cu succes, însă pe un pacient s-a constatat o progresie a
11
scoliozei.
Deși se pare că rezultatele tratamentului sunt benefice, au fost semnalate câteva cazuri de
deces sponan la pacienții care erau în curs de terapie, însă nu s-a stabilit cu exactitate dacă
terapia în sine a avut o legătură directă cu acesta. [19] Una dintre posibilele cauze ale decesului ar
fi o disfuncție respiratorie datorată afectării hipotalamusului, complicație care ar fi legată de
genele afectate în acest sindrom, ce duc au ca sinteză chiar hipotalamusul.
B.3.2. Tratament hormonal la adulți
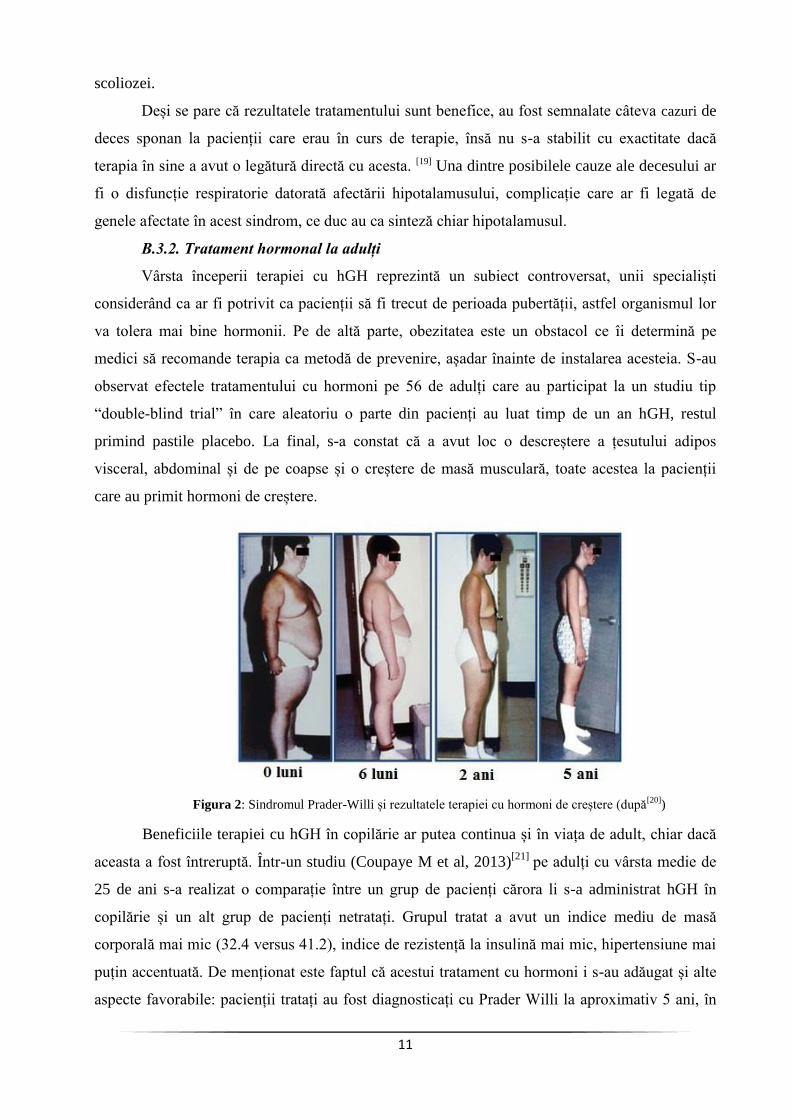
Vârsta începerii terapiei cu hGH reprezintă un subiect controversat, unii specialiști
considerând ca ar fi potrivit ca pacienții să fi trecut de perioada pubertății, astfel organismul lor
va tolera mai bine hormonii. Pe de altă parte, obezitatea este un obstacol ce îi determină pe
medici să recomande terapia ca metodă de prevenire, așadar înainte de instalarea acesteia. S-au
observat efectele tratamentului cu hormoni pe 56 de adulți care au participat la un studiu tip
“double-blind trial” în care aleatoriu o parte din pacienți au luat timp de un an hGH, restul
primind pastile placebo. La final, s-a constat că a avut loc o descreștere a țesutului adipos
visceral, abdominal și de pe coapse și o creștere de masă musculară, toate acestea la pacienții
care au primit hormoni de creștere.
Figura 2: Sindromul Prader-Willi și rezultatele terapiei cu hormoni de creștere (după[20]
)
Beneficiile terapiei cu hGH în copilărie ar putea continua și în viața de adult, chiar dacă
aceasta a fost întreruptă. Într-un studiu (Coupaye M et al, 2013)[21]
pe adulți cu vârsta medie de
25 de ani s-a realizat o comparație între un grup de pacienți cărora li s-a administrat hGH în
copilărie și un alt grup de pacienți netratați. Grupul tratat a avut un indice mediu de masă
corporală mai mic (32.4 versus 41.2), indice de rezistență la insulină mai mic, hipertensiune mai
puțin accentuată. De menționat este faptul că acestui tratament cu hormoni i s-au adăugat și alte
aspecte favorabile: pacienții tratați au fost diagnosticați cu Prader Willi la aproximativ 5 ani, în

12
Figura 3: Producerea de distomie uniparentală (după [29]
)
comparație cu grupul netratat la care vârsta medie este de 10 ani.
Au fost, totodata, studiate diferite subtipuri de Prader Willi în funcție de cauza apariției
sindromului și, astfel, s-a ajuns la ideea conform căreia subiecțiicare prezintă deleții la nivelul
cromozomului 15 răspund mai bine la tratamentul cu hGH decât pacienții cu disomia
uniparentală.[22]
În ceea ce privește efectele adverse ale tratamentului cu hGH au fost semnalate scăderi
ale nivelului de insulină, progresie a scoliozei, edem la nivelul extremităților[23]
, probleme
respiratorii.
Cunoscând faptul ca pacienții cu Prader Willi sunt predispuși la diabet zaharat[24]
, este
importantă o atentă monitorizare a acestora pe parcursul tratamentului. Alterarea metabolismului
glucozei se referă la faptul că terapia cu hGH poate duce, în unele cazuri, la o rezistență crescută
la insulină datorită efectului invers obținut în mod obișnuit pe acțiunea insulinică.
O altă posibilă complicație este scolioza, survenită ca o asociere între obezitate și
hipotonie musculară, care a fost remarcată la 70% din pacienții unui studiu[25]
, atât în grupul de
control, cât și în cel de tratament[26]
. Scolioza afectează de la 30% până la 80% dintre pacienții
cu Prader-Willi. Multiple studii au evidențiat faptul că terapia în cauză nu produce nici un efect
asupra scoliozei, mai mult decât atât scolioza nu este considerată o contraindicație în inițierea
sau continuarea terapiei cu hGH.[13]
Atât la adulți, cât și la copii, au fost semnalate anormalități la nivelul sistemului respirator
și un potențial crescut de apariție de sindrom de apnee obstructivă în somn (OSA)[27,28]
prin
asocierea de: tonus muscular scăzut, obezitate, hipoxie. Posibilitatea apriției acestei complicații
se datorează creșterii nivelului de IGF-1 în timpul administrării de hGH și acest lucru poate duce
la hiperplazie limfoidă.
Toate aceste efecte adverse menționate sunt un risc al terapiei cu hGH, iar acest risc
trebuie analizat atent în balanță cu beneficiile, fiecare pacient având un tablou clinic unic și
complex.
III. DISCUȚII ȘI CONCLUZII
III.A. DISCUȚII
A.1. Amprentarea genomică și
disomia uniparentală
Cauzele apariției sindomului Prader Willi
au fost îndelung studiate de cercetători, iar
disomia uniparentală (UPD) și amprentarea
genomică (imprinting genes) sunt două

13
dintre acestea. Defectul se produce de la fecundație, odată cu formarea celulei-ou și în consecință
fătul va avea două copii ale aceluiași cromozom (matern sau patern) sau ale unei secvențe de pe
cromozomul respectiv. În multe cazuri această eroare nu are niciun efect asupra sănătății sau
dezvoltării individului, însă în altele, cum ar fi sindromul Prader Willi sau sindromul Angelman,
pacienții dezvoltă patologii grave ce le afectează calitatea vieții.[29]
Procesul de amprentare, numit și proces de metilare, se petrece într-un procent mic și
marchează, dar și deosebește genele moștenite de la mamă de cele de la tată. Uneori, genele
amprentate se găsesc grupate în loci apropiați ca de exemplu pe brațul p al cromozomului 11
(sindromul Beckwith-Wiedemann) sau brațul q al cromozomului 15 (sindroamele Prader Willi,
Angelman).
A.2.Comportamentul obsesiv-compulsiv
O mare parte din persoanele care au sindromul Prader Willi dezvoltă pe parcursul vieții
dermatilomanie, un impuls incontrolabil de a-și leza tegumentul, dar și comportament obsesiv-
compulsiv, în prescurtare: OCD[30]
. Tratamentul standard pentru aceste tulburări includ terapia
cognitiv-comportamentală și, după caz, antidepresive: fluoxetine, fluvoxamine sau sertraline.
Din nefericire, datorită problemelor de vorbire și retardului mental variat, acești pacienți
cooperează cu dificultate cu psihoterapeuții, așadar complianța la tratament este scăzută. Un
“avantaj” al acestui comportament îl constituie faptul că pacienții vor avea tendința obsesivă de a
respecta o rutină zilnică, prin urmare li se poate recomanda un program cu mese la ore fixe, sport
și kinetoterapie, având un efect de potențial control asupra obezității și îmbunătățire a calității
vieții.
A.3. Durata tratamentului
În ceea ce privește durata de administrare a terapiei cu hormoni de creștere, cel mai lung
studiu ca durată compară un grup de copii cu vârste cuprinse între 6 și 9 ani - care au fost tratați
cu hormoni de creștere pentru 6 ani începând de la 4-20 de luni de viață - cu un grup de copii de
aceeași vârstă și sex, dar care nu au urmat tratament. Rezultatele au relevat faptul că cei care
aparțineau grupului care primea tratament au prezentat o scădere semnificativă a grăsimii
corporale, o masă musculară crescută, profile lipidice mult mai favorabile și o putere și funcție
motorie mult mai bune.[31]
A.4. Prognostic și rata de mortalitate
Morbiditatea
în rândul pacienților cu Prader-Willi are o rată crescută, obezitatea
caracteristică acestor pacienți fiind factorul principal. Acestuia i se adaugă și complicațiile
apărute pe parcursul vieții, dar și dizabilitatea intelectuală care aduce o creștere a ratei
morbidității de 1.7 ori față de populația generală[32]
.

14
Pacienţii cu sindrom Prader Willi ajung adesea la vârsta adultă dar, datorită complexităţii
afecţiunii, necesită îngrijiri particulare şi multidisciplinare.[33]
Speranța de viață medie este de
aproximativ 30 de ani (în România aceasta este vârsta maximă atinsă), însă este important de
menționat faptul că este posibil ca aceasta să crească datorită faptului ca actualele generații de
pacienți cu Prader Willi au avut posibilitatea de a opta pentru tratamentul cu hGH.
Au fost raportate câteva cazuri de deces subit de când se utilizează tratamentul cu
hormoni de creștere uman. Un studiu realizat în acest sens a comparat informațiile cu privire la
decesul neașteptat al pacienților care au primit sau nu tratament cu hGH (13 pacienți decedați -
grupul A, de la 9 luni la 34 de ani, care nu au urmat terapia, respectiv 7 pacienți decedați care au
fost tratați cu hGH- grupul B). După ce a fost comparată cauza decesurilor între cele două
grupuri s-a constatat faptul că moartea survenită la cei care au urmat tratament hormonal nu este
esențial diferită față de cei care nu au urmat terapia. Rezultatele studiului au indicat faptul că
ceea ce stă la baza decesului acestora este reprezentat de respirația dereglată și disfuncția
hipotalamică, cause ce pot fi exarcebate de terapia cu hGH. Această concluzie duce la o mai
mare atenție acordată de către clinicieni a determinării dozei de hGH și a monitorizării condiției
respiratorii a pacienților atunci când li se acordă acest tratament.[34]
III. B. CONCLUZII- IDEI
1. Sindromul Prader Willi este principala cauză de obezitate sindromică, printre
patologiile asociate frecvente observate diabetul zaharat, hiperfagie, apnee, deficit statural, edem
periferic și mai rar: scolioză, hipotiroidism, deficit de hormoni suprarenalieni.
2. Din punct de vedere cognitiv-comportamental, pacienții suferă de tulburări obsesiv-
compulsive, dermatilomanie, retard mintal moderat, aceste probleme aducând și mai multe
complicații în tabloul clinic.
3. Cauzele apariției acestui sindrom sunt: în cea mai mare parte delețiile (până la 70%),
disomiile uniparentale (10-15%) și amprentare genomica.
4. Diagnosticarea precoce este un element-cheie pentru prevenirea complicațiilor.
5. Tratamentul medicamentos standard, aprobat și recomandat în domeniul medical, este
reprezentat de terapia cu hormoni de creștere umani (hGH) care aduce avantaje precum:
creșterea în înălțime și a masei musculare, scăderea procentului de țesut adipos subcutanat și
visceral, dezvoltarea capacităților intelectuale, dar și dezavantaje: tulburări de somn, apnee,
predispoziție la diabet, edem, accentuarea scoliozei.
6. Nu se poate aprecia cu exactitate durata administrării terapiei cu hGH, aceasta
depinzând și de vârsta la care a debutat, dar și de răspunsul pacientului la tratament.

15
7. Studiile arată ca începerea terapiei din perioada copilăriei are efecte benefice mai
pronunțate, efecte care s-ar părea ca pot rămâne pe termen lung chiar și după încheierea
tratamentului.
8. Deși tratamentul nu este fără risc, beneficiile pot duce la o îmbunătățire a calității vieții
și la scăderea morbidității persoanelor suferind de Prader-Willi. Specialiștii recomandă începerea
administrării de hGH cât mai precoce, dar sub o atentă monitorizare a clinicianului.

16
IV. BIBLIOGRAFIE
1. Merlin Butler, Management of Prader-Willi Syndrome 2006, SUA
2. Jill E Emerick, Karen S Vogt; Endocrine manifestations and management of Prader-Willi
syndrome; Emerick and Vogt International Journal of Pediatric Endocrinology 2013.
3. Cassidy SB, Schwartz S; Prader-Willi and Angelman syndromes. Disorders of genomic
imprinting; Department of Genetics, Case Western Reserve University, Cleveland, Ohio, USA.
4. http://www.ncbi.nlm.nih.gov/books/NBK1330/figure/pws.F2/?report=objectonly
5. Suzanne B. Cassady, E. Dykens, Charles A. Williams; 2 Prader-Willi and Angelman
Syndromes: Sister Imprinted Disorders; American journal of medical genetics (Semin. Med.
Genet.) 97:136–146 (2000).
6. W F Paterson, M D C Donaldson: Growth hormone therapy in the Prader-Willi syndrome.
Arch Dis Child 2003;88:283-285 doi:10.1136/adc.88.4.283
7. http://www.nature.com/gim/journal/v14/n1/images/gim0b013e31822bead0t1.gif
8. Munce T, Simpson R, Bowling F: Molecular characterization of Prader-Willi syndrome by
real-time PCR. Genet Test. 2008 Jun;12(2):319-24. doi: 10.1089/gte.2007.0105.
9. Daniel J Driscoll, Jennifer L Miller, Stuart Schwartz, Suzanne B Cassidy, Prader-Willi
Syndrome
10. E. Braha, A. Sireteanu, C. Vulpoi, C. Gorduza, D. Branisteanu, R. Popescu1, C. Badiu, C.
Rusu; Clinical and endocrine aspects of five Prader Willi patients; “Grigore T. Popa” University
of Medicine and Pharmacy - 1 Department of Medical Genetics – 2 Department of
Endocrinology, 3 “Sf. Spiridon” Hospital Outpatient Unit - Department of Endocrinology, Iasi, 4
“Carol Davila” University of Medicine and Pharmacy - Department of Endocrinology,
Bucharest, Romania
11. Deal CL, Tony M, Hoybye C, Allen DB, Tauber M, Christiansen JS, Growth Hormone in
Prader-Willi Syndrome Clinical Care Guidelines Workshop P, Collaboration E: Growth hormone
research society workshop summary: consensus guidelines for recombinant human growth
hormone therapy in prader-willi syndrome. J Clinic Endocrinol and metab 2013, 98(6):E1072–
E1087.
12. Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M: Recommendations
for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab 2008,
93(11):4183–4197.

17
13. Miller JL: Approach to the child with prader-willi syndrome. J Clin Endocrinol Metab 2012,
97(11):3837–3844.
14. Burman P, Ritzen EM, Lindgren AC: Endocrine dysfunction in Prader-Willi syndrome: a
review with special reference to GH. Endocrine reviews 2001, 22(6):787–799.
15. Diene G, Mimoun E, Feigerlova E, Caula S, Molinas C, Grandjean H, Tauber M: Endocrine
disorders in children with Prader-Willi syndrome–data from 142 children of the French database.
Hormone Res Paediat 2010, 74(2):121–128.
16. Deal CL, Tony M, Hoybye C, Allen DB, Tauber M, Christiansen JS, Growth Hormone in
Prader-Willi Syndrome Clinical Care Guidelines Workshop P, Collaboration E: Growth hormone
research society workshop summary: consensus guidelines for recombinant human growth
hormone therapy in prader-willi syndrome. J Clinic Endocrinol and metab 2013, 98(6):E1072–
E1087.
17. Eiholzer U, L'allemand D, Schlumpf M, Rousson V, Gasser T, Fusch C :Growth hormone
and body composition in children younger than 2 years with Prader-Willi syndrome. J Pediatr.
2004 Jun;144(6):753-8.
18. Myers SE, Whitman BY, Carrel AL, Moerchen V, Bekx MT, Allen DB :Two years of
growth hormone therapy in young children with Prader-Willi syndrome: physical and
neurodevelopmental benefits. Am J Med Genet A. 2007 Mar 1;143A(5):443-8.
19. Nagai T, Obata K, Tonoki H, Temma S, Murakami N: Cause of sudden, unexpected death of
Prader-Willi syndrome patients with or without growth hormone treatment. Am J Med Genet A.
2005 Jul 1;136(1):45-8.
20. http://lookfordiagnosis.com/mesh_info.php?term=prader-willi+syndrome&lang=1
21. Coupaye M, Lorenzini F, Lloret-Linares C, et al: Growth hormone therapy for children and
adolescents with Prader-Willi syndrome is associated with improved body composition and
metabolic status in adulthood. J Clinic Endocrinol and metab 2013, 98(2):E328–E335.
22. Grugni G, Giardino D, Crinò A, Malvestiti F, Ballarati L, Di Giorgio G, Marzullo P: Growth
hormone secretion among adult patients with Prader-Willi syndrome due to different genetic
subtypes. J Endocrinol Invest 2011, 34(7):493–497.
23. Sanchez-Ortiga R, Klibanski A, Tritos N.A: Effects of recombinant human growth hormone
therapy in adults with Prader-Willi syndrome: a meta-analysis. Clin Endocrinol (Oxf). 2012
Jul;77(1):86-93.
24. Butler, J.V.; Whittington, J.E.; Holland, A.J.; Boer, H.; Clarke, D.; Webb, T. Prevalence of,
and risk factors for, physical ill-health in people with prader-willi syndrome: A population-based
study. Dev. Med. Child Neurol. 2002, 44, 248–255.

18
25. Carrel AL, Myers SE, Whitman BY, Allen DB: Growth hormone improves body
composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome:
A controlled study. J Pediatr. 1999 Feb;134(2):215-21.
26. Burman P, Ritzen E. Martin, Lindgren C: Endocrine dysfunction in Prader Willi Syndrome:
A review with a special reference to GH. Endocrine Reviews 2002 Jan; 22(6):787-799
27. Miller J, Silverstein J, Shuster J, Driscoll DJ, Wagner M: Short-term effects of growth
hormone on sleep abnormalities in Prader-Willi syndrome. J Clin Endocrinol Metab. 2006
Feb;91(2):413-7. Epub 2005 Nov 29.
28. Sedky K, Bennett DS, Pumariega A: Prader Willi syndrome and obstructive sleep apnea: co-
occurrence in the pediatric population. J Clin Sleep Med. 2014 Apr 15;10(4):403-9. doi:
10.5664/jcsm.3616.
29. http://www.bjmg.edu.mk/record.asp?subrecordid=582
30. Shaffer Lisa G., Agan Noelle: American College of Medical Genetics Statement on
Diagnostic Testing for Uniparental Disomy. Genet Med. 2001 May-Jun; 3(3): 206–211
31. Clarke D.J, Boer H., Whittington J., Holland A., Butler J., Webb T.: Prader -Willi syndrome,
compulsive and ritualistic behaviours: the first population-based survey. The British Journal of
Psychiatry Apr 2002, 180 (4) 358-362; DOI: 10.1192/bjp.180.4.358
32. Carrel AL, Myers SE, Whitman BY, Eickhoff J, Allen DB: Long-term growth hormone
therapy changes the natural history of body composition and motor function in children with
Prader-Willi syndrome. J Clinic Endocrinol and metab 2010, 95(3):1131–1136.
33. Einfeld S, Kavanagh S, Smith A, Evans E, Tonge B, Taffe J: Mortality in Prader-Willi
Syndrome. Am J Ment Retard. 2006 May; 111(3): 193–198. doi: 10.1352/0895-
8017(2006)111[193:MIPS]2.0.CO;2
34. Dr. Maria Puiu, Dr. Cristina Skrypnyk; Mic GHID DE DIAGNOSTIC ÎN BOLILE RARE;
Sinteza materialului „Esenţialul în 101 Boli genetice rare”; Editura Victor Babeş 2009, pg. 15
35. T. Nagai, K. Obata, H. Tonoki, S. Temma, et al: Cause of Sudden, Unexpected Death of
Prader–Willi Syndrome Patients With or Without Growth Hormone Treatment; American
Journal of Medical Genetics, 2005, 136A:45–48
http://www.ncbi.nlm.nih.gov/pubmed/?term=Driscoll%20DJ%5BAuthor%5D&cauthor=true&cauthor_uid=16317059


















