
9. Deficiențe enzimatice · Catalazei umane (eritrocite) Situs de legare catalaza umana Kirkman &...
56
1250 boli genetice 200 erori metabolice deficiente enzimatice 9. Deficiențe enzimatice 9.1. Boli corelate cu deficiențe enzimatice și proteice
Transcript of 9. Deficiențe enzimatice · Catalazei umane (eritrocite) Situs de legare catalaza umana Kirkman &...

1250 boli genetice
200 erori metabolice
deficiente enzimatice
9. Deficiențe enzimatice
9.1. Boli corelate cu deficiențe enzimatice și proteice
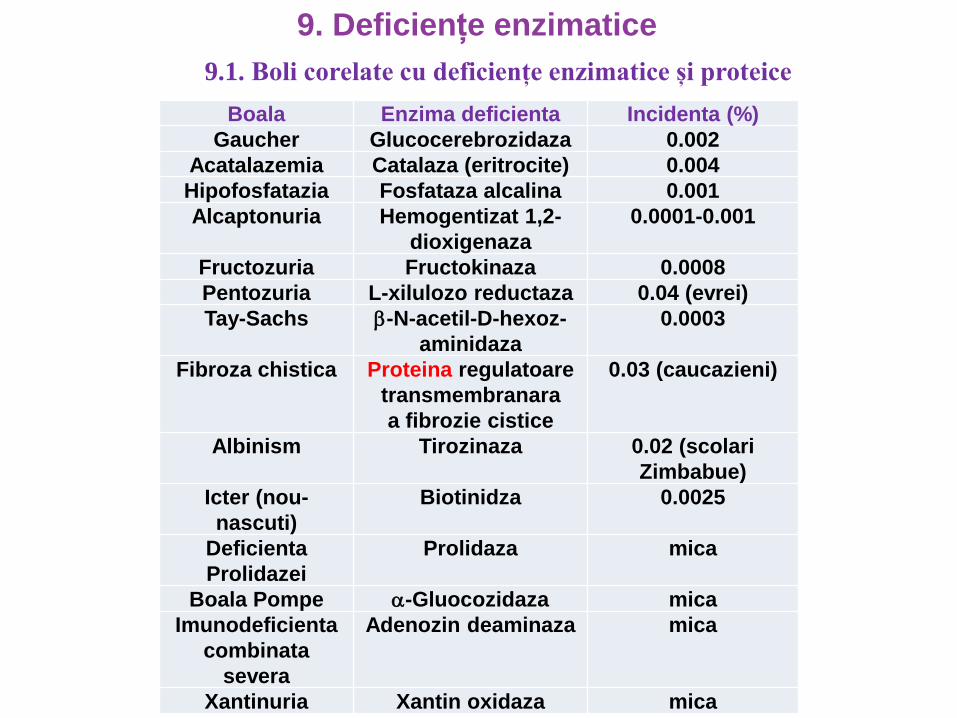
Boala Enzima deficienta Incidenta (%)
Gaucher Glucocerebrozidaza 0.002
Acatalazemia Catalaza (eritrocite) 0.004
Hipofosfatazia Fosfataza alcalina 0.001
Alcaptonuria Hemogentizat 1,2-
dioxigenaza
0.0001-0.001
Fructozuria Fructokinaza 0.0008
Pentozuria L-xilulozo reductaza 0.04 (evrei)
Tay-Sachs -N-acetil-D-hexoz-
aminidaza
0.0003
Fibroza chistica Proteina regulatoare
transmembranara
a fibrozie cistice
0.03 (caucazieni)
Albinism Tirozinaza 0.02 (scolari
Zimbabue)
Icter (nou-
nascuti)
Biotinidza 0.0025
Deficienta
Prolidazei
Prolidaza mica
Boala Pompe -Gluocozidaza mica
Imunodeficienta
combinata
severa
Adenozin deaminaza mica
Xantinuria Xantin oxidaza mica
9. Deficiențe enzimatice
9.1. Boli corelate cu deficiențe enzimatice și proteice
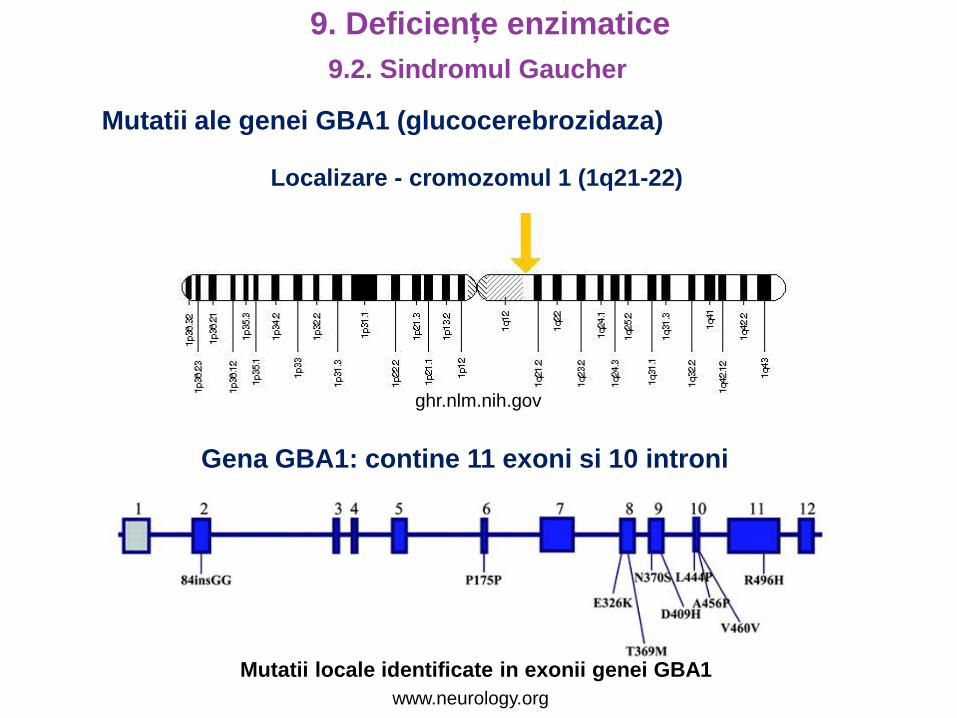
9.2. Sindromul Gaucher
Mutatii ale genei GBA1 (glucocerebrozidaza)
Localizare - cromozomul 1 (1q21-22)
Gena GBA1: contine 11 exoni si 10 introni
ghr.nlm.nih.gov
www.neurology.org
Mutatii locale identificate in exonii genei GBA1
9. Deficiențe enzimatice
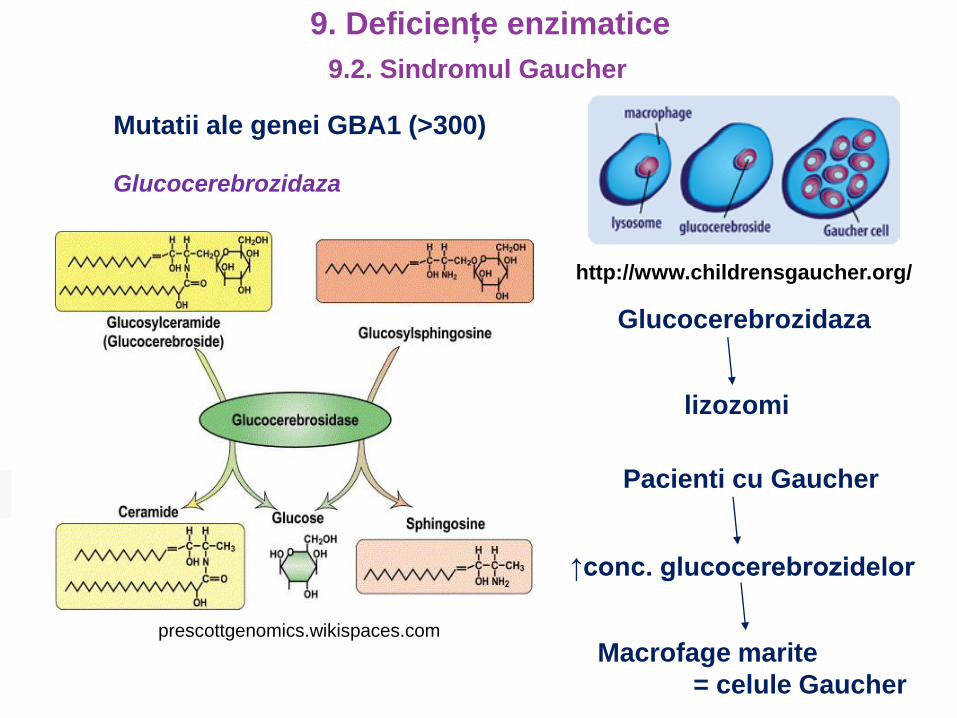
Glucocerebrozidaza
Mutatii ale genei GBA1 (>300)
prescottgenomics.wikispaces.com
http://www.childrensgaucher.org/
Glucocerebrozidaza
lizozomi
Pacienti cu Gaucher
↑conc. glucocerebrozidelor
Macrofage marite
= celule Gaucher
9.2. Sindromul Gaucher
9. Deficiențe enzimatice
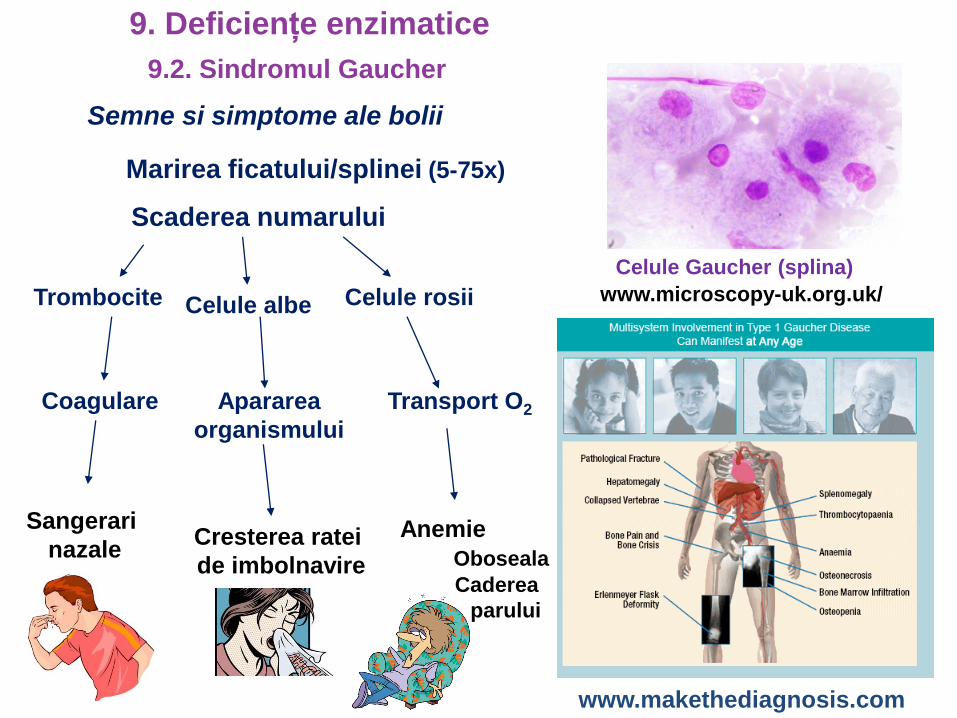
Semne si simptome ale bolii
Marirea ficatului/splinei (5-75x)
Scaderea numarului
Trombocite Celule albe Celule rosii
www.makethediagnosis.com
Celule Gaucher (splina)
www.microscopy-uk.org.uk/
Coagulare
Sangerari
nazale
Apararea
organismului
Cresterea ratei
de imbolnavire
Transport O2
Anemie
Oboseala
Caderea
parului
9.2. Sindromul Gaucher
9. Deficiențe enzimatice
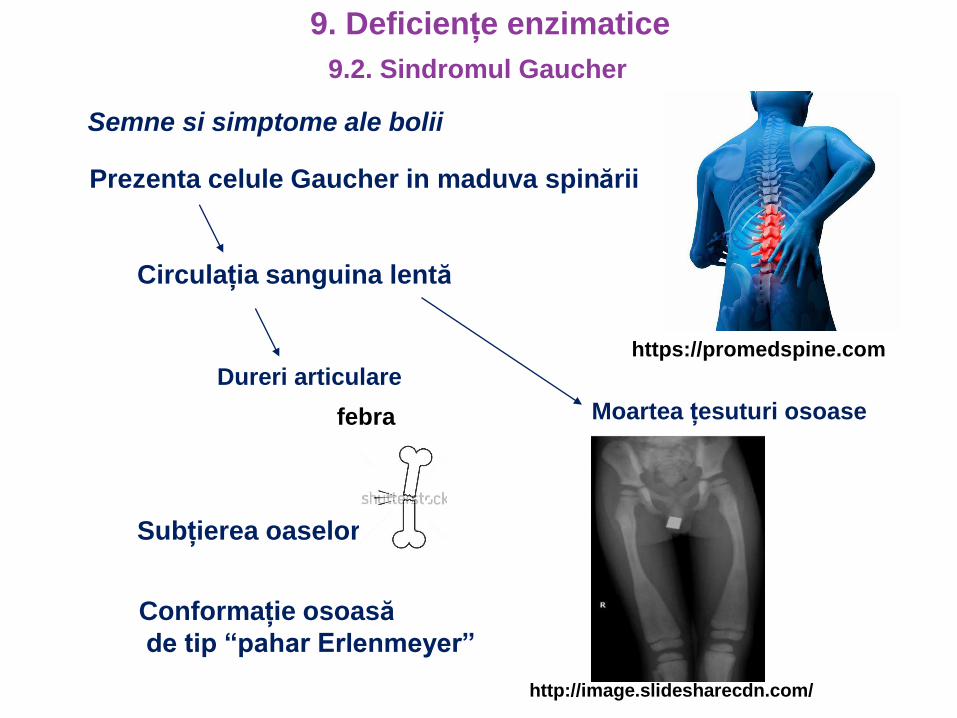
Subțierea oaselor
Conformație osoasă
de tip “pahar Erlenmeyer”
Semne si simptome ale bolii
Prezenta celule Gaucher in maduva spinării
Circulația sanguina lentă
Dureri articulare
https://promedspine.com
febra Moartea țesuturi osoase
http://image.slidesharecdn.com/
9.2. Sindromul Gaucher
9. Deficiențe enzimatice
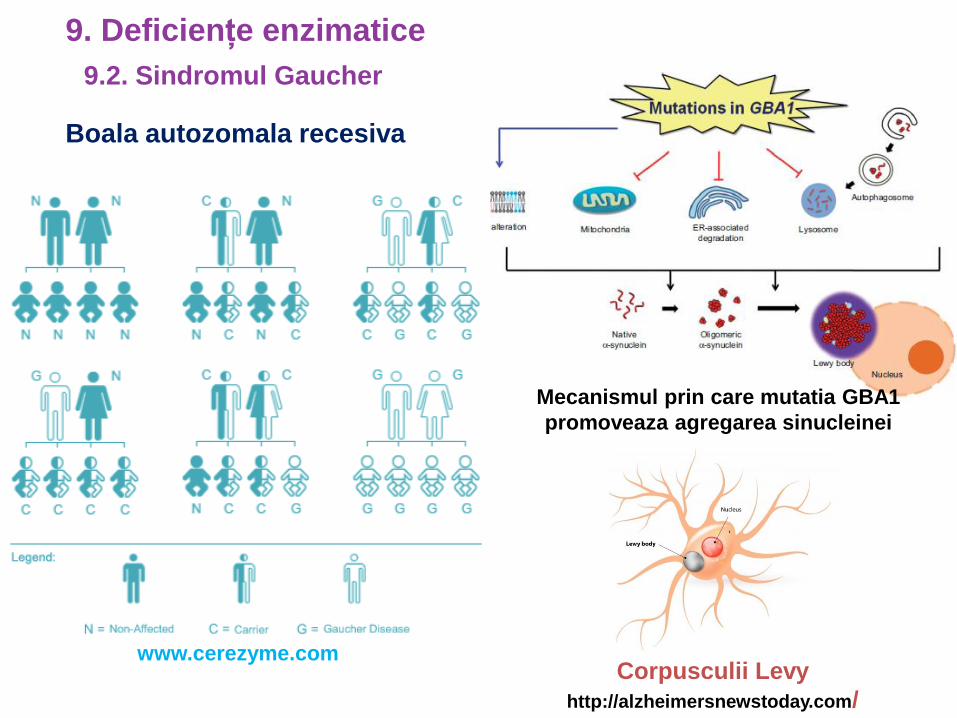
Boala autozomala recesiva
www.cerezyme.com
Mecanismul prin care mutatia GBA1
promoveaza agregarea sinucleinei
Corpusculii Levy
http://alzheimersnewstoday.com/
9.2. Sindromul Gaucher
9. Deficiențe enzimatice
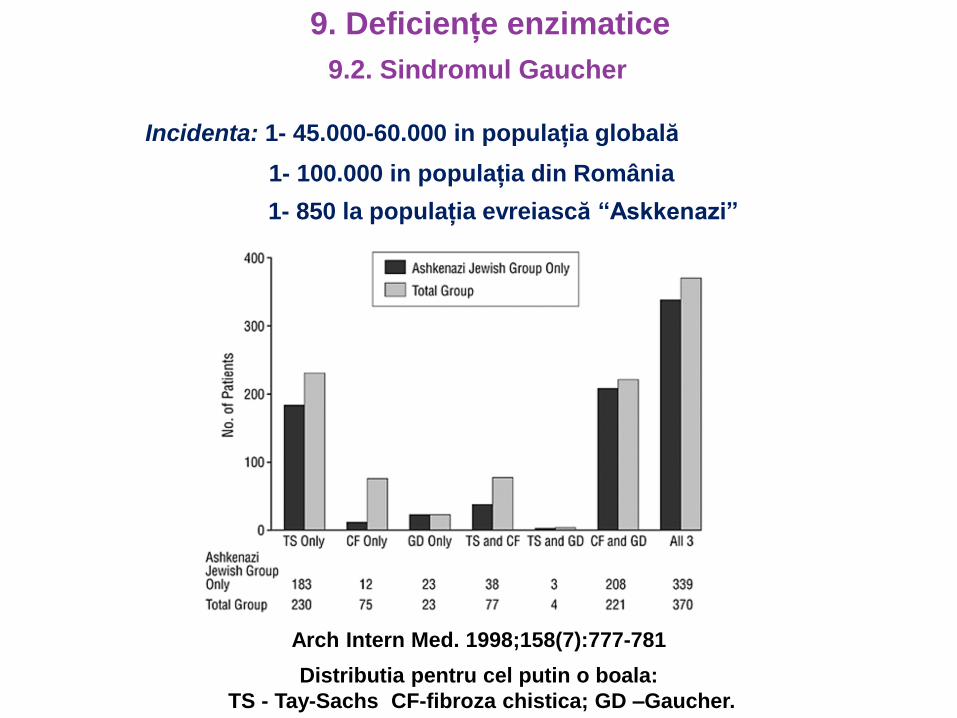
Incidenta: 1- 45.000-60.000 in populația globală
1- 850 la populația evreiască “Askkenazi”
Arch Intern Med. 1998;158(7):777-781
Distributia pentru cel putin o boala:
TS - Tay-Sachs CF-fibroza chistica; GD –Gaucher.
1- 100.000 in populația din România
9.2. Sindromul Gaucher
9. Deficiențe enzimatice
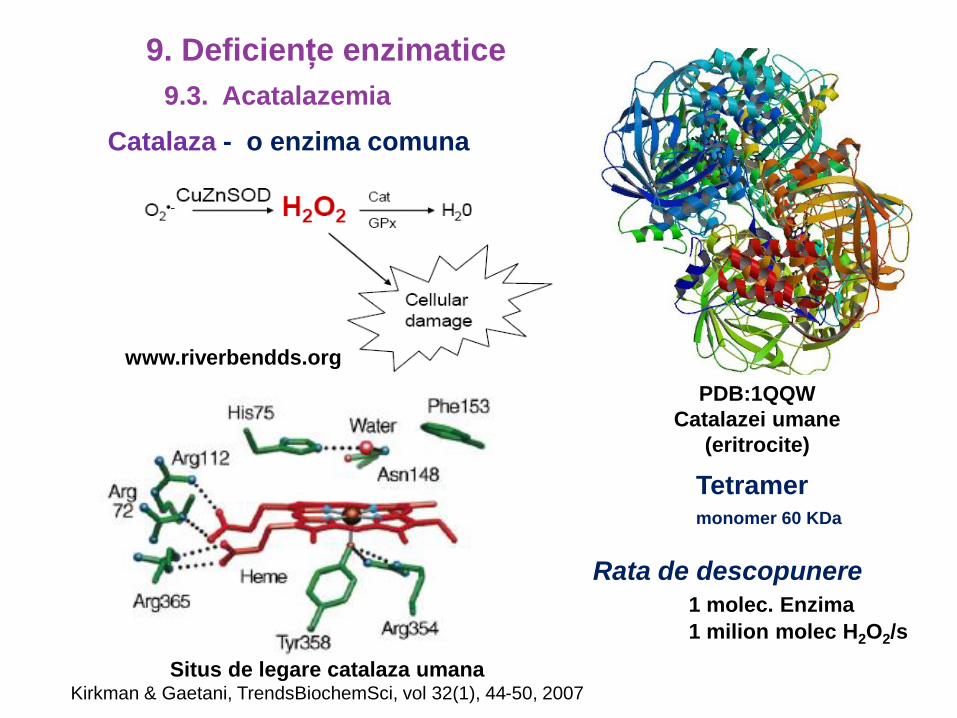
Catalaza - o enzima comuna
www.riverbendds.org
Rata de descopunere
1 molec. Enzima
1 milion molec H2O2/s
PDB:1QQW
Catalazei umane
(eritrocite)
Situs de legare catalaza umanaKirkman & Gaetani, TrendsBiochemSci, vol 32(1), 44-50, 2007
Tetramermonomer 60 KDa
9.3. Acatalazemia
9. Deficiențe enzimatice
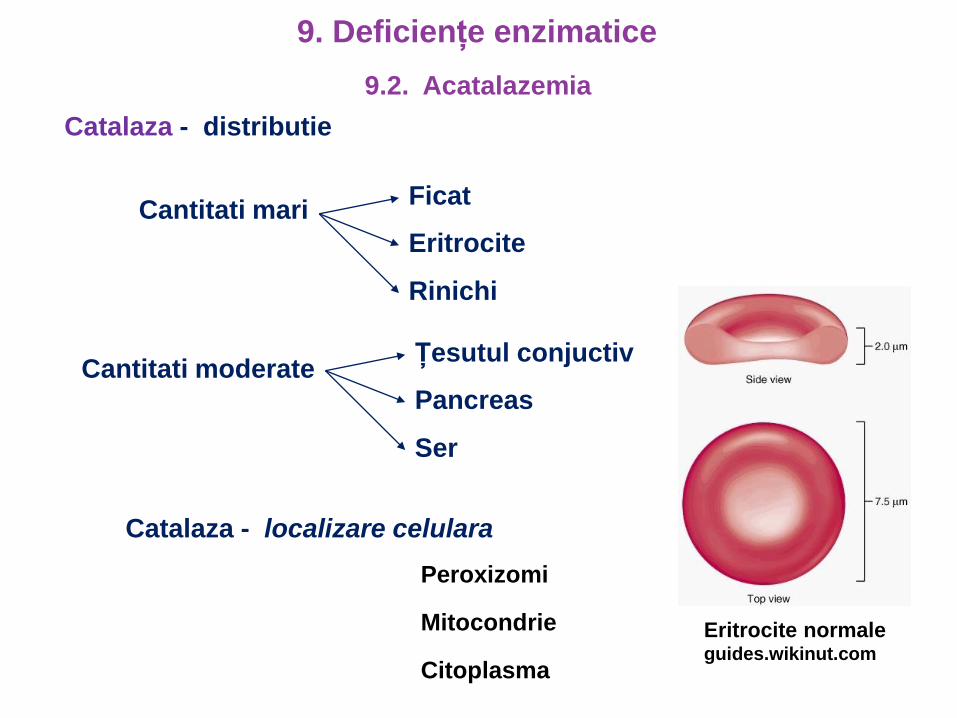
Catalaza - distributie
Cantitati mariFicat
Eritrocite
Rinichi
Cantitati moderateȚesutul conjuctiv
Pancreas
Ser
Catalaza - localizare celulara
Peroxizomi
Citoplasma
Mitocondrie Eritrocite normaleguides.wikinut.com
9.2. Acatalazemia
9. Deficiențe enzimatice
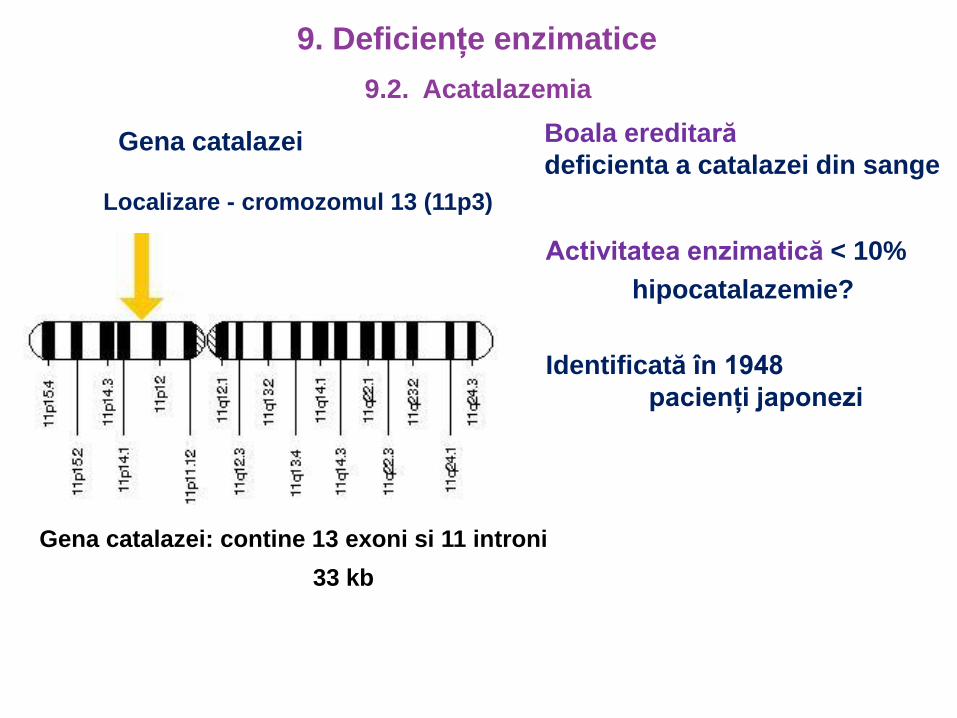
Gena catalazei
Localizare - cromozomul 13 (11p3)
Gena catalazei: contine 13 exoni si 11 introni
33 kb
9.2. Acatalazemia
9. Deficiențe enzimatice
Boala ereditară
deficienta a catalazei din sange
Activitatea enzimatică < 10%
hipocatalazemie?
Identificată în 1948
pacienți japonezi

9.2. Acatalazemia
9. Deficiențe enzimatice
Incidenta: 0,05- 1000 în populația ungară (homozigot)
2,3/1000 la în populația ungară (heterozigot)
118 cazuri de acatalazemie
61 familii (11țări)
11 mutații
8 în exoni
3 în introni
1 :12 500 în populația niponă
0,04- 1000 în populația elvețiană
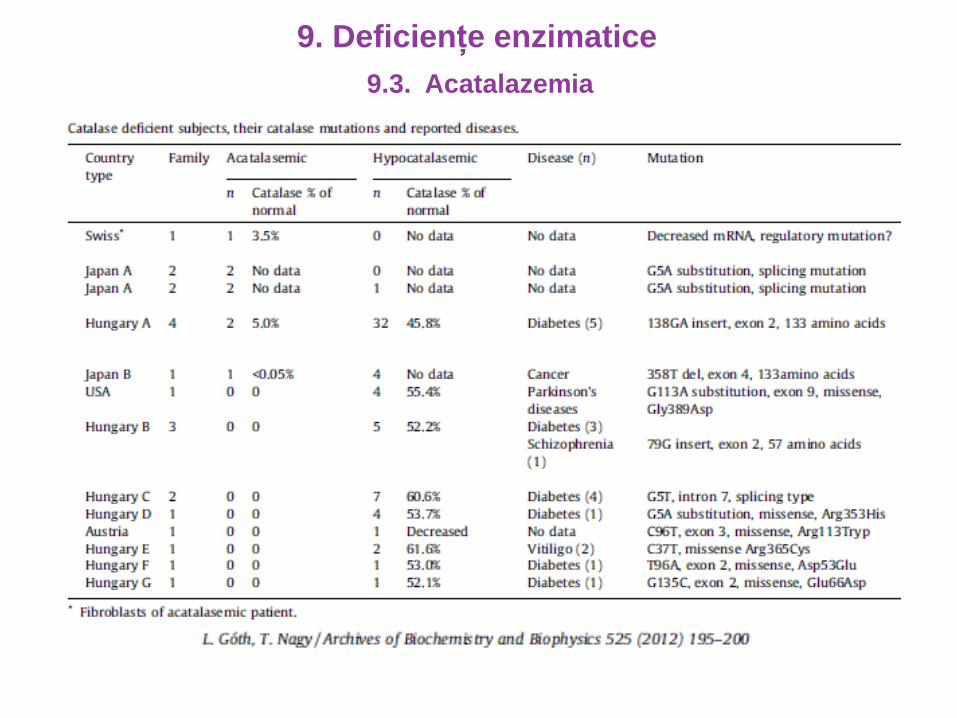
9.3. Acatalazemia
9. Deficiențe enzimatice
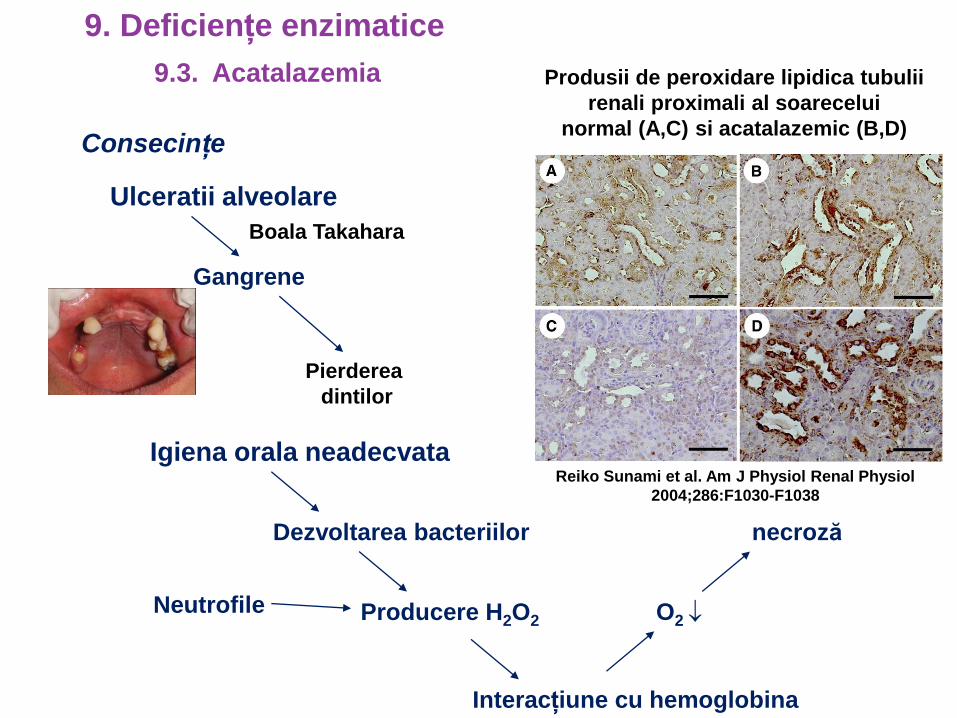
Consecințe
Ulceratii alveolare
Gangrene
Pierderea
dintilor
Igiena orala neadecvata
Dezvoltarea bacteriilor
Producere H2O2
Interacțiune cu hemoglobina
O2
necroză
Reiko Sunami et al. Am J Physiol Renal Physiol
2004;286:F1030-F1038
Produsii de peroxidare lipidica tubulii
renali proximali al soarecelui
normal (A,C) si acatalazemic (B,D)
Neutrofile
9.3. Acatalazemia
9. Deficiențe enzimatice
Boala Takahara
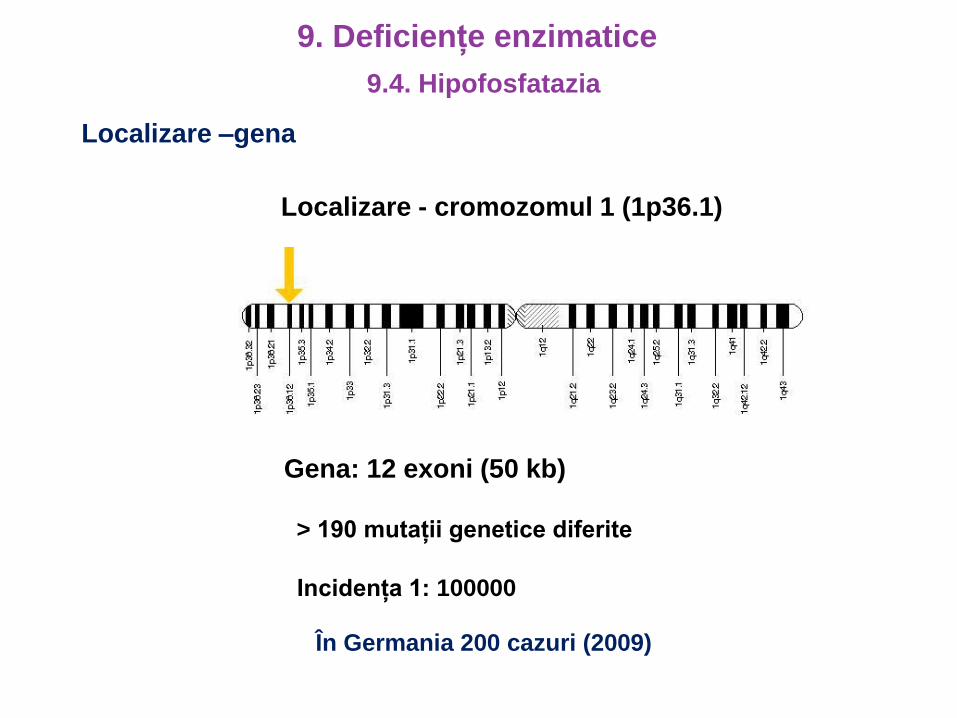
Localizare –gena
Localizare - cromozomul 1 (1p36.1)
Gena: 12 exoni (50 kb)
9.4. Hipofosfatazia
9. Deficiențe enzimatice
> 190 mutații genetice diferite
Incidența 1: 100000
În Germania 200 cazuri (2009)
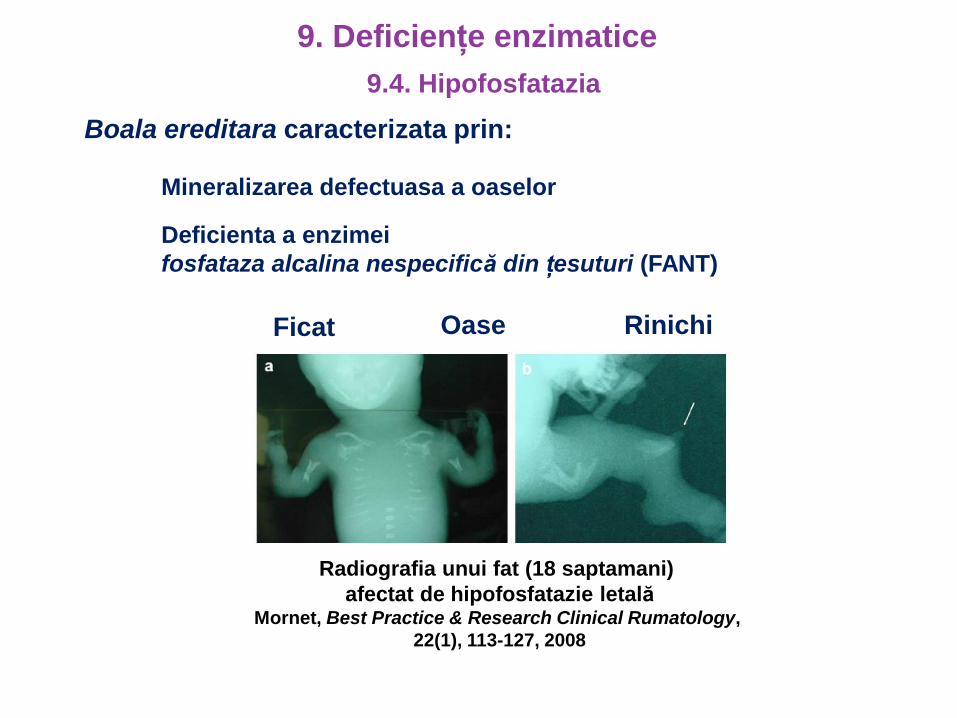
Boala ereditara caracterizata prin:
Mineralizarea defectuasa a oaselor
Deficienta a enzimei
fosfataza alcalina nespecifică din țesuturi (FANT)
Ficat Oase Rinichi
Radiografia unui fat (18 saptamani)
afectat de hipofosfatazie letalăMornet, Best Practice & Research Clinical Rumatology,
22(1), 113-127, 2008
9.4. Hipofosfatazia
9. Deficiențe enzimatice
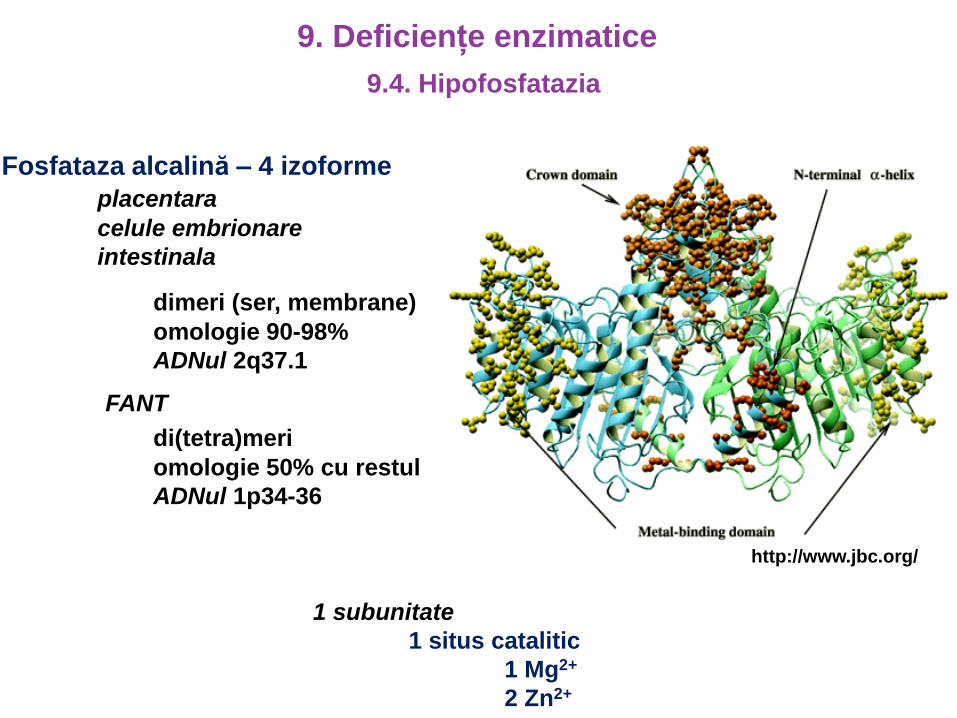
Fosfataza alcalină – 4 izoforme
placentara
celule embrionare
intestinala
dimeri (ser, membrane)
omologie 90-98%
ADNul 2q37.1
FANT
di(tetra)meri
omologie 50% cu restul
ADNul 1p34-36
http://www.jbc.org/
1 subunitate
1 situs catalitic
1 Mg2+
2 Zn2+
9.4. Hipofosfatazia
9. Deficiențe enzimatice
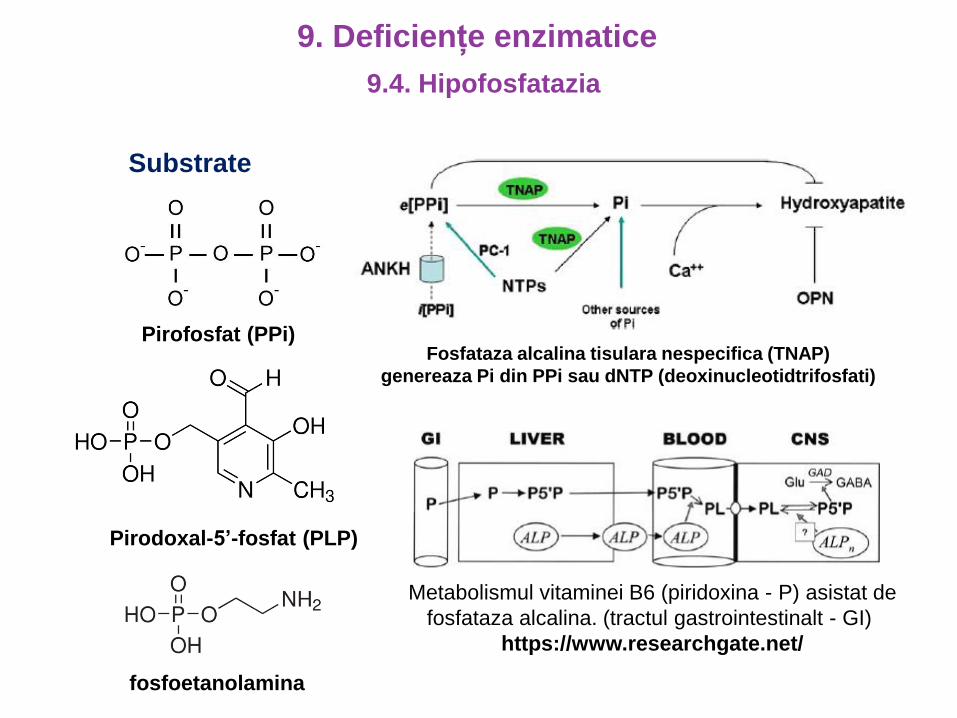
Substrate
Fosfataza alcalina tisulara nespecifica (TNAP)
genereaza Pi din PPi sau dNTP (deoxinucleotidtrifosfati)
Pirofosfat (PPi)
Pirodoxal-5’-fosfat (PLP)
fosfoetanolamina
Metabolismul vitaminei B6 (piridoxina - P) asistat de
fosfataza alcalina. (tractul gastrointestinalt - GI)
https://www.researchgate.net/
9.4. Hipofosfatazia
9. Deficiențe enzimatice

Mutatii genetice
(>190)
Mornet, Best Practice & Research
Clinical Rumatology, 22(1), 113-127, 2008
c.1559delT 41%
F310L 14%
populatia japoneza
c.571G>A 50%
(E174K )
Populatia europeana
(hipofosfatalazemie
moderata)
9.4. Hipofosfatazia
9. Deficiențe enzimatice
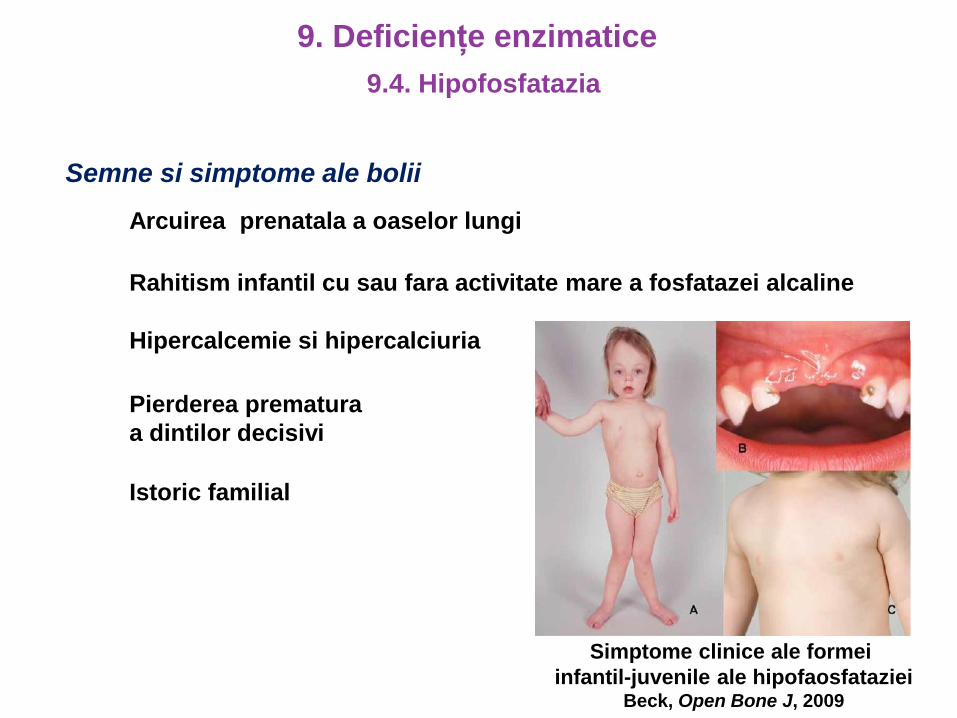
Semne si simptome ale bolii
Arcuirea prenatala a oaselor lungi
Rahitism infantil cu sau fara activitate mare a fosfatazei alcaline
Hipercalcemie si hipercalciuria
Pierderea prematura
a dintilor decisivi
Istoric familial
Simptome clinice ale formei
infantil-juvenile ale hipofaosfatazieiBeck, Open Bone J, 2009
9.4. Hipofosfatazia
9. Deficiențe enzimatice

Valori de referinta folosite pentru
diagnoza HP la copii/adulti
Metode de diagnosticate
Densitatea osoasa femurala
Pirodoxal-5’-fosfat (PLP)
Marker al hipofosfataziei
9.4. Hipofosfatazia
9. Deficiențe enzimatice

www.netterimages.com
Deficienta a -N-acetil-D-hexoz-aminidazei
Enzima - scindeaza gangliozidele GM2
GM2 – glicosfingolipide (acid sialic)
Abundente in plasma neuronilor
Deficienta acumularea GM2
in lizozomii celulelor nervoase
www.stockphotos.ro
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice

Incidenta:
1- 27/1-30 la evreii Ashkenazi
1- 110 la evreii marocani
1- 140 la evreii irachieini
1- 50 la americanii irlandezi
1- 250 in populatia generala
Boala recesiva
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice
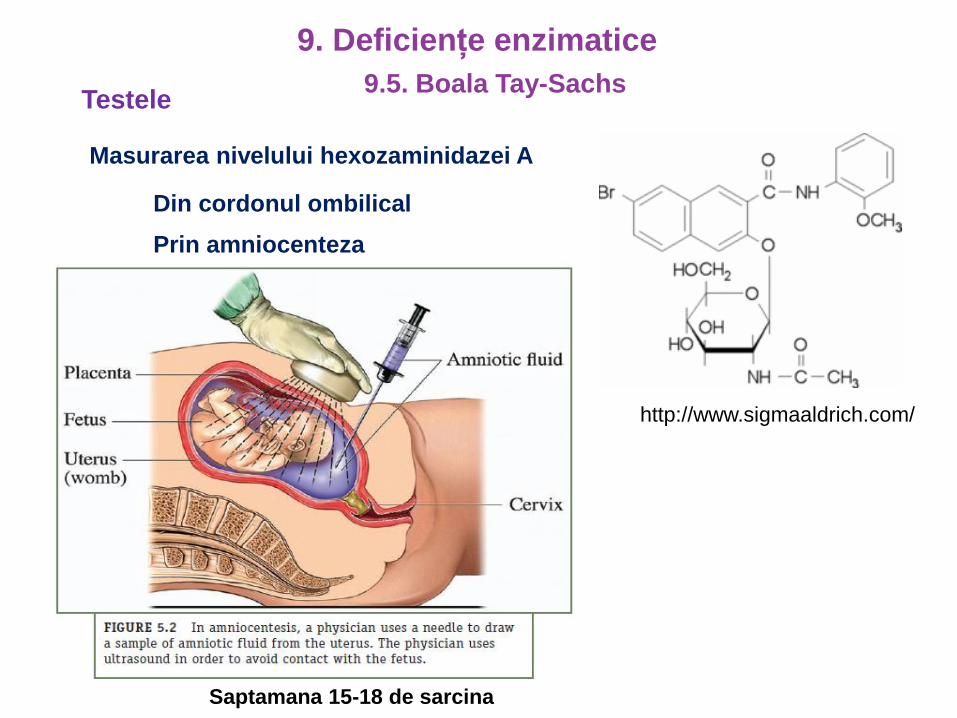
Saptamana 15-18 de sarcina
Testele
Masurarea nivelului hexozaminidazei A
Din cordonul ombilical
Prin amniocenteza
http://www.sigmaaldrich.com/
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice
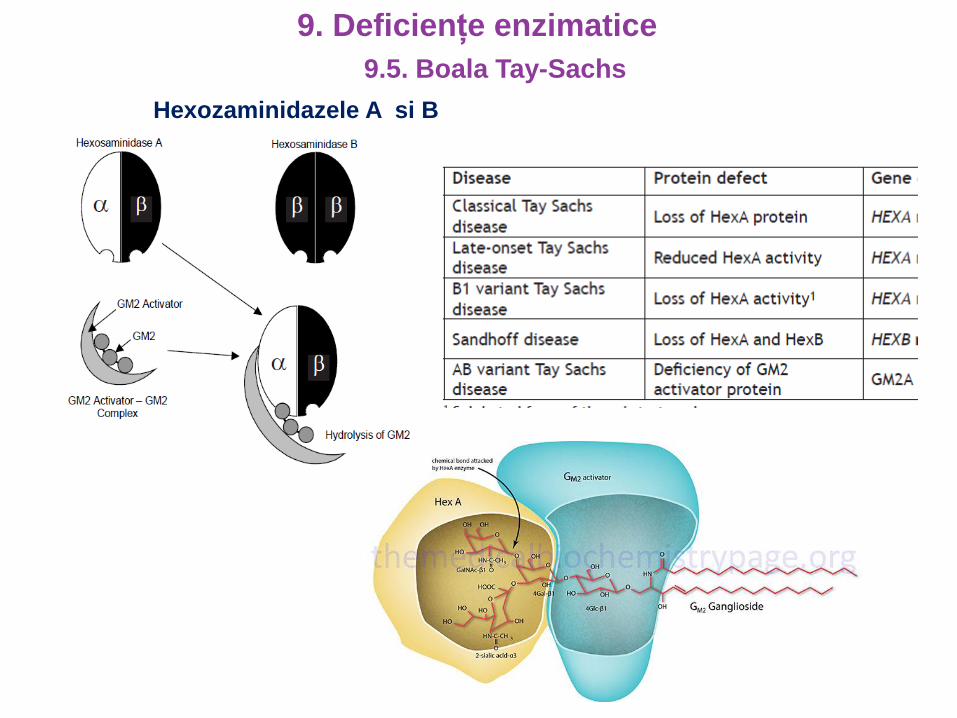
Hexozaminidazele A si B
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice
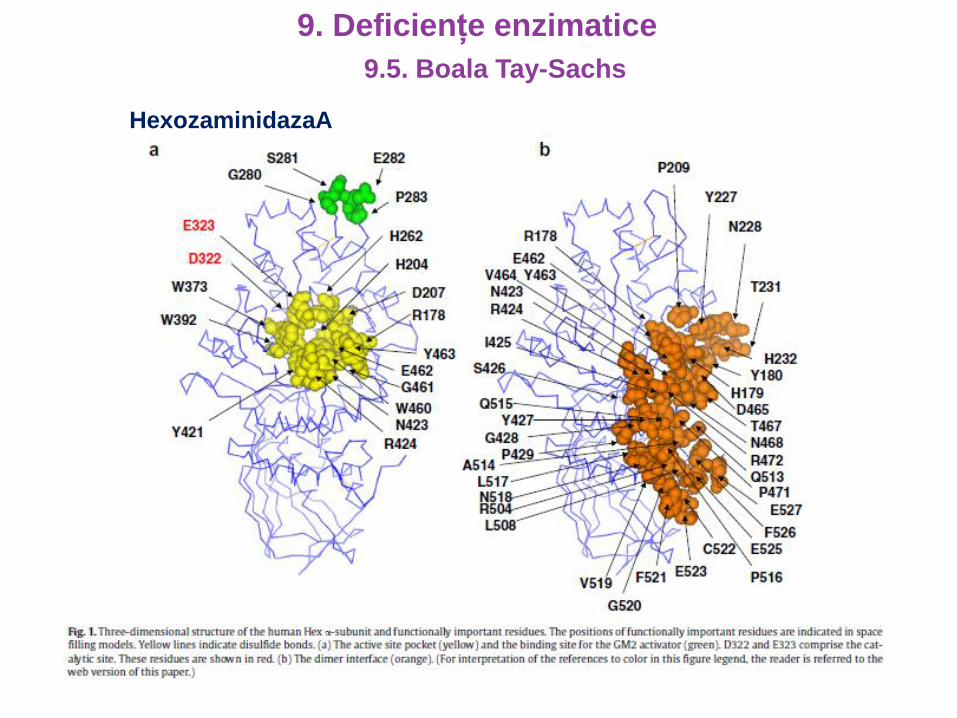
HexozaminidazaA
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice

HexozaminidazaA
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice

Retard mental (primele 4-8 luni)
Descreste atentia vizuala (6-10 luni)
Gangliozidoza cronica GM2 - adolescenti
tremurat
coordinare slaba
tulburari de vorbire
retina neafectata
Manifestari
Boala Tay-Sachs infantila (deficienta hexozaminidazei A)
Deficiente motorii medii (3-6 luni)
Descrec miscarile voluntare si capacitatea vocala
(>10 luni)
Stare vegetativa, orb (2 ani) decesul (4 ani)
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice

Boala Sanhoff- mutatii ale gene HexB
codeaza ambele subunitati HexA si HexB
infantilă, juvenilă sau târzie
Manifestări
Boala Tay-Sachs –varianta B1
Enzima își reduce activitatea față de unele substrate
Apare la 3-7 ani
Deteriorarea este variabila
deces 1-2 ani
> 5 ani
Pete maculare rosii
http://themedicalbiochemistrypage.org/
9.5. Boala Tay-Sachs
9. Deficiențe enzimatice
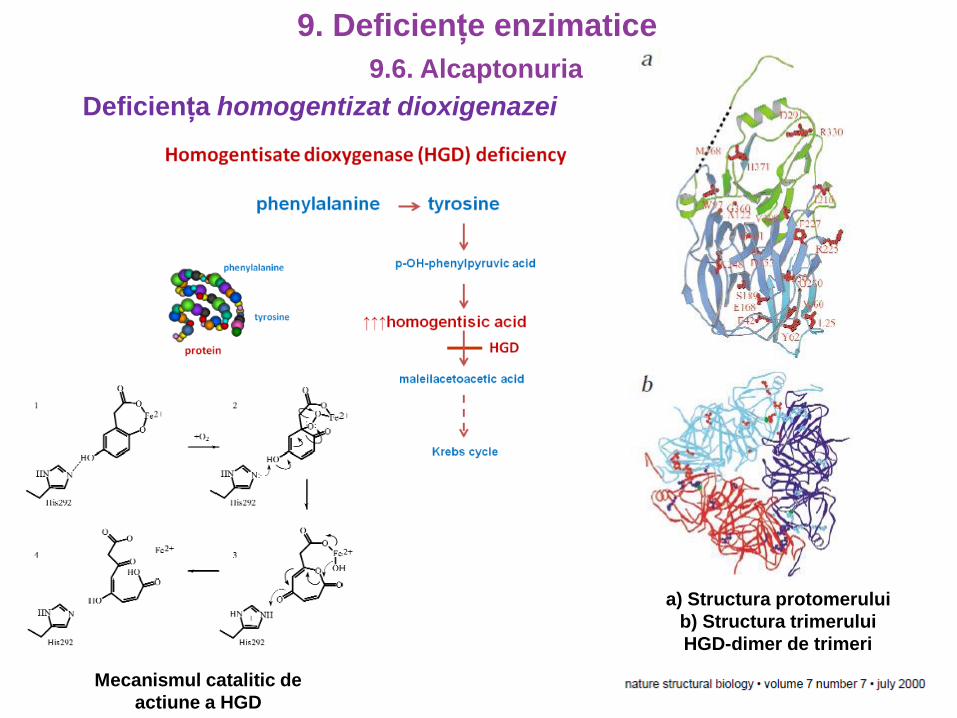
Deficiența homogentizat dioxigenazei
a) Structura protomerului
b) Structura trimerului
HGD-dimer de trimeri
Mecanismul catalitic de
actiune a HGD
9.6. Alcaptonuria
9. Deficiențe enzimatice
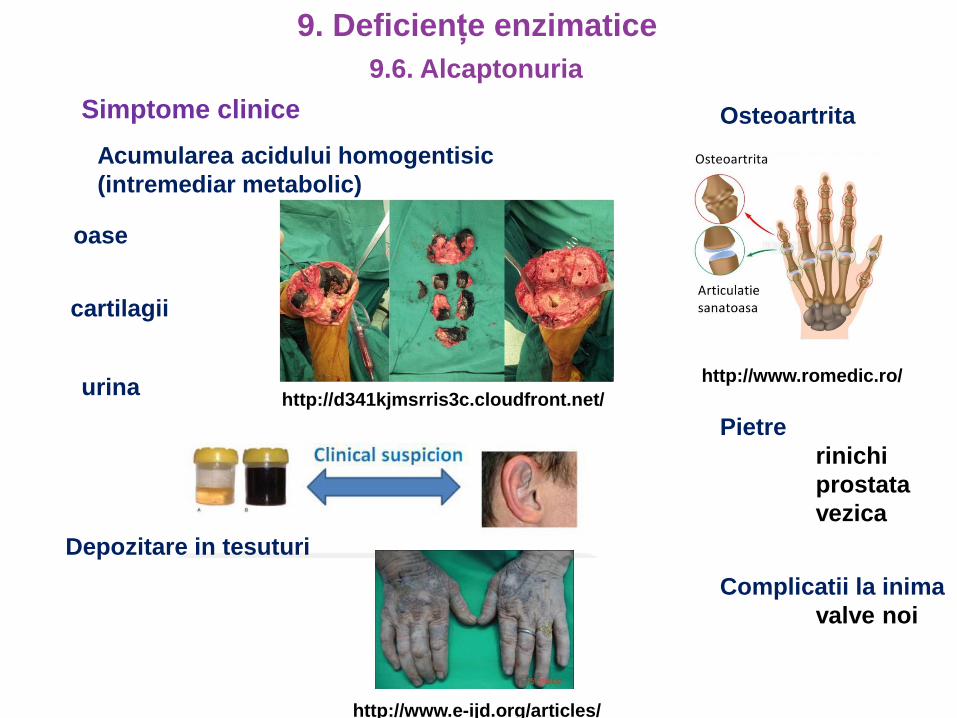
Simptome clinice
Acumularea acidului homogentisic
(intremediar metabolic)
oase
cartilagii
urinahttp://d341kjmsrris3c.cloudfront.net/
Depozitare in tesuturi
http://www.e-ijd.org/articles/
Osteoartrita
http://www.romedic.ro/
Pietre
rinichi
prostata
vezica
Complicatii la inima
valve noi
9.6. Alcaptonuria
9. Deficiențe enzimatice
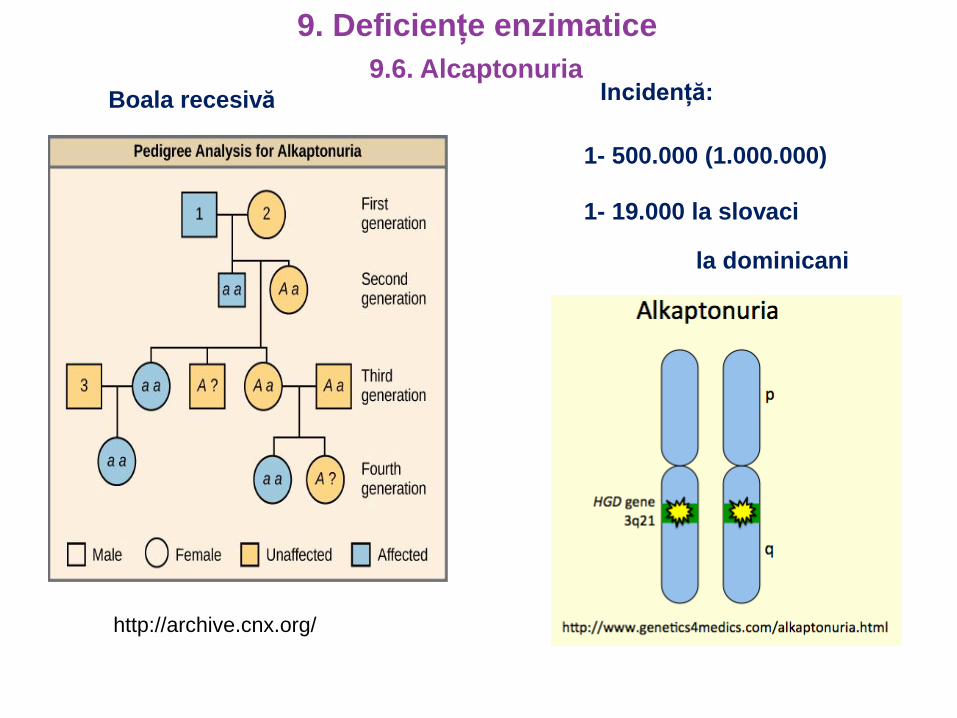
Boala recesivă
http://archive.cnx.org/
Incidență:
1- 500.000 (1.000.000)
1- 19.000 la slovaci
la dominicani
9.6. Alcaptonuria
9. Deficiențe enzimatice
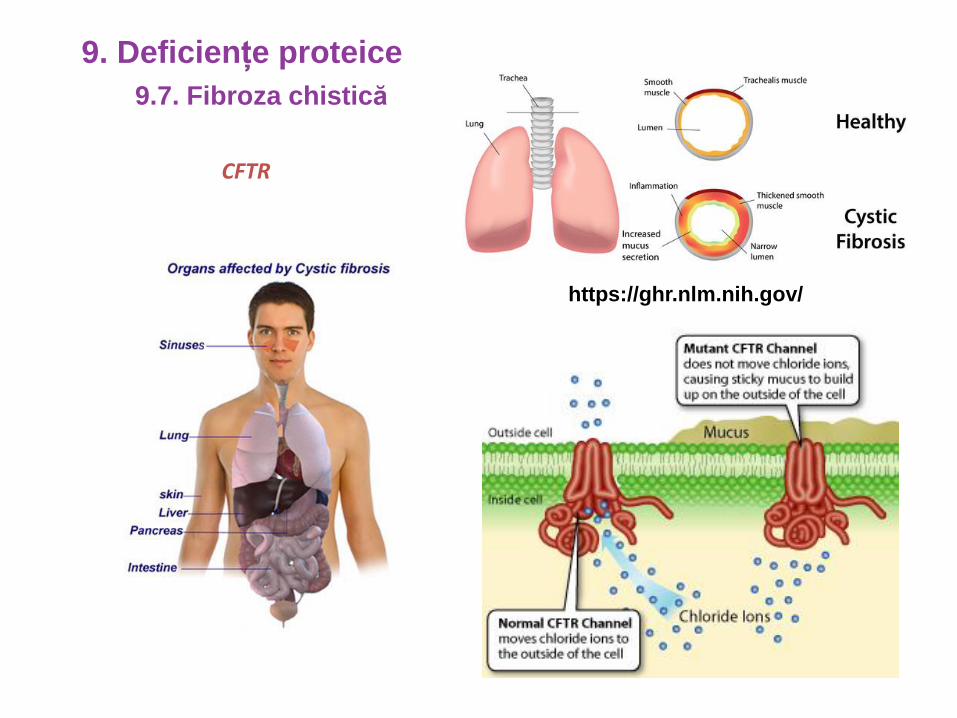
CFTR
https://ghr.nlm.nih.gov/
9.7. Fibroza chistică
9. Deficiențe proteice
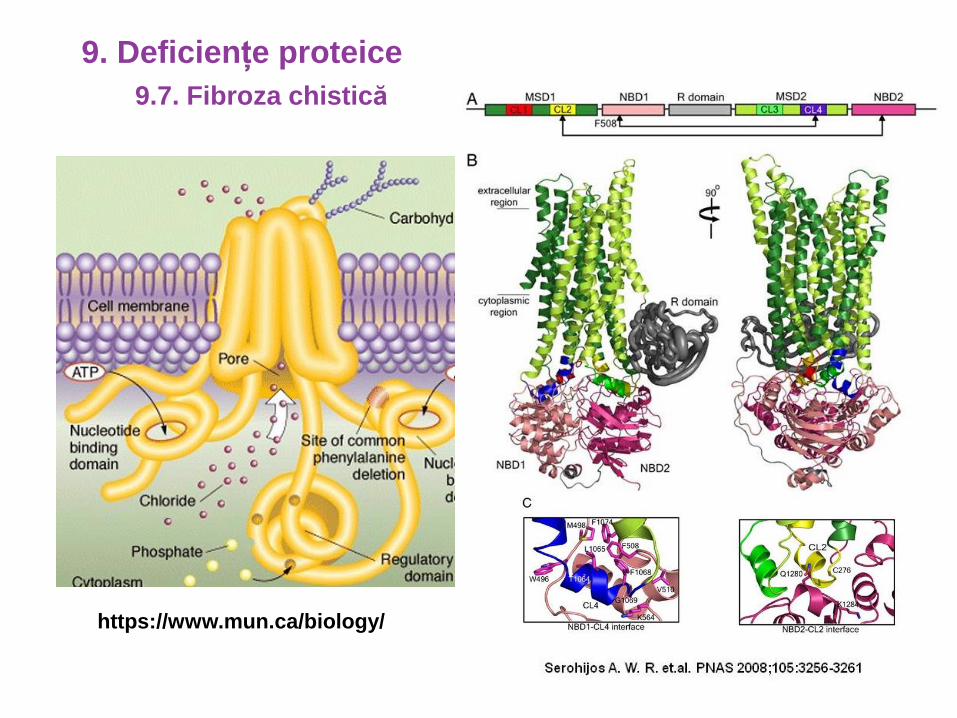
https://www.mun.ca/biology/
9.7. Fibroza chistică
9. Deficiențe proteice
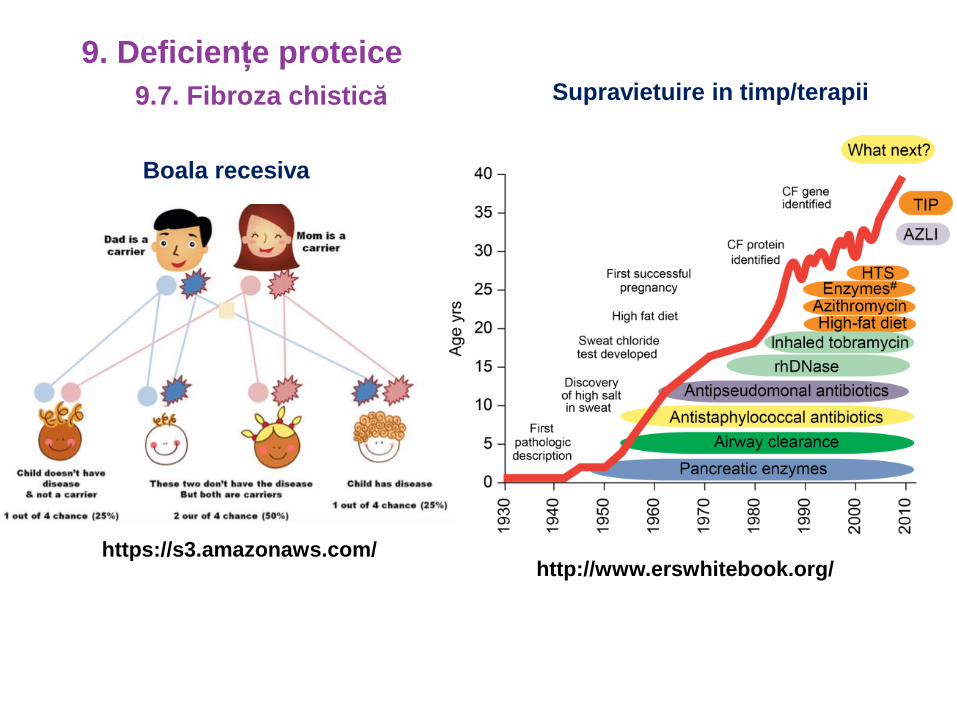
Boala recesiva
Supravietuire in timp/terapii
https://s3.amazonaws.com/http://www.erswhitebook.org/
9.7. Fibroza chistică
9. Deficiențe proteice
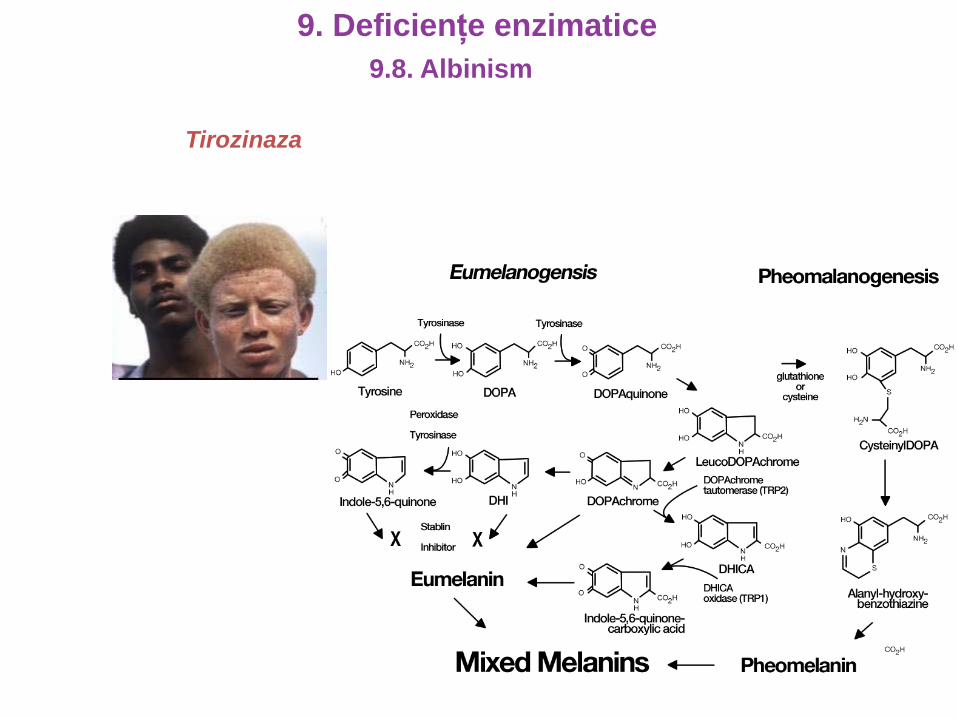
Tirozinaza
9.8. Albinism
9. Deficiențe enzimatice

Terminologie
Pigmentare redusă (piele, păr, ochi)
https://www.epharmapedia.com
9.8. Albinism
9. Deficiențe enzimatice
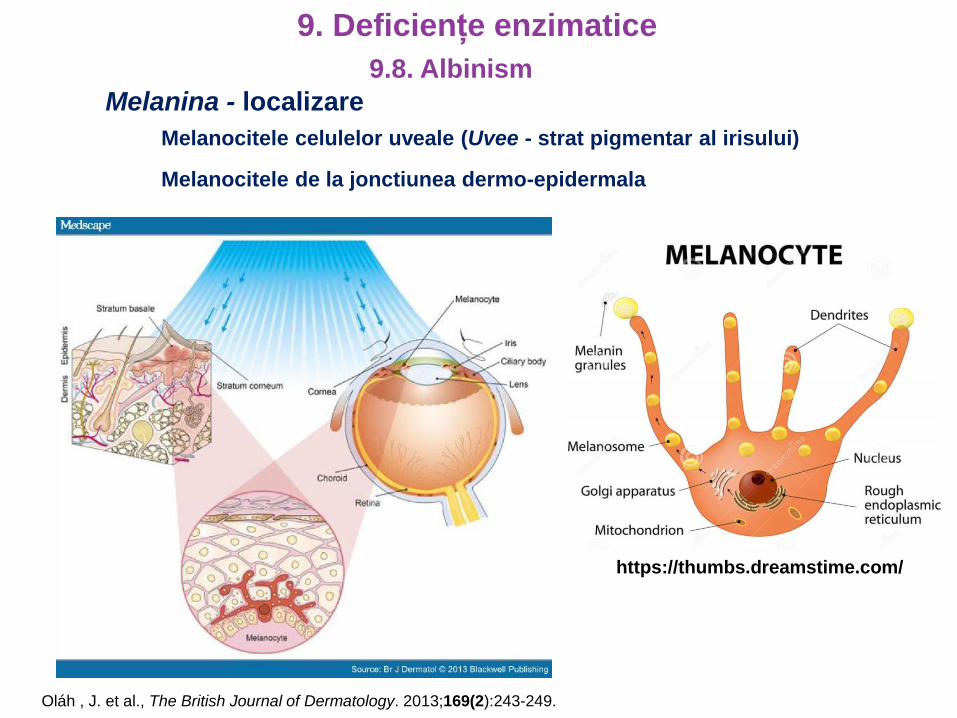
Melanina - localizare
Melanocitele celulelor uveale (Uvee - strat pigmentar al irisului)
Oláh , J. et al., The British Journal of Dermatology. 2013;169(2):243-249.
Melanocitele de la jonctiunea dermo-epidermala
https://thumbs.dreamstime.com/
9.8. Albinism
9. Deficiențe enzimatice
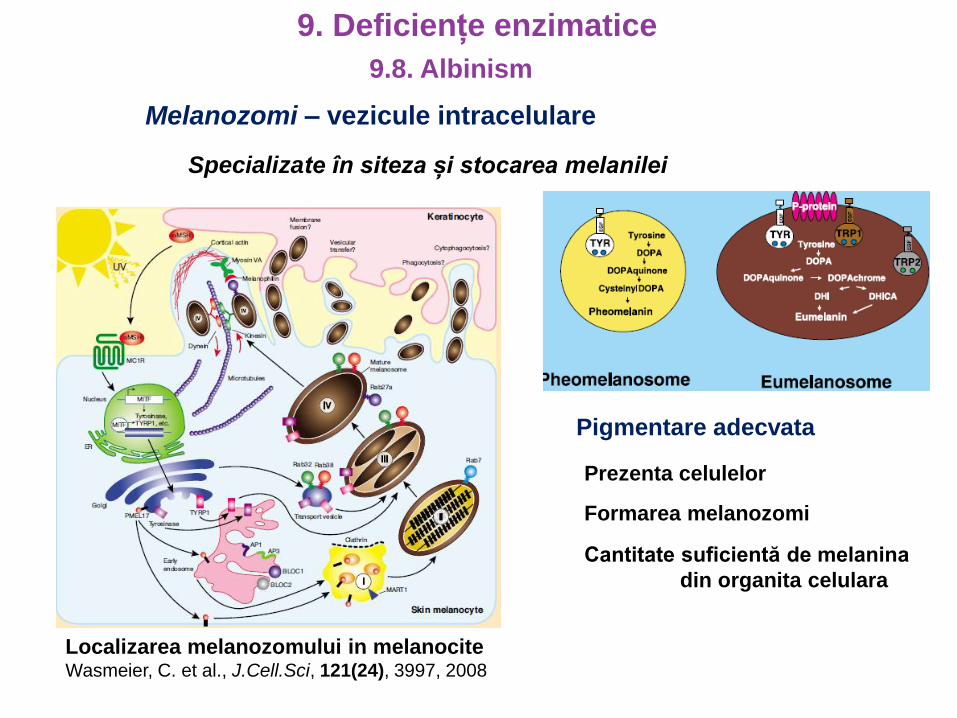
Melanozomi – vezicule intracelulare
Specializate în siteza și stocarea melanilei
Localizarea melanozomului in melanociteWasmeier, C. et al., J.Cell.Sci, 121(24), 3997, 2008
Pigmentare adecvata
Prezenta celulelor
Formarea melanozomi
Cantitate suficientă de melanina
din organita celulara
9.8. Albinism
9. Deficiențe enzimatice
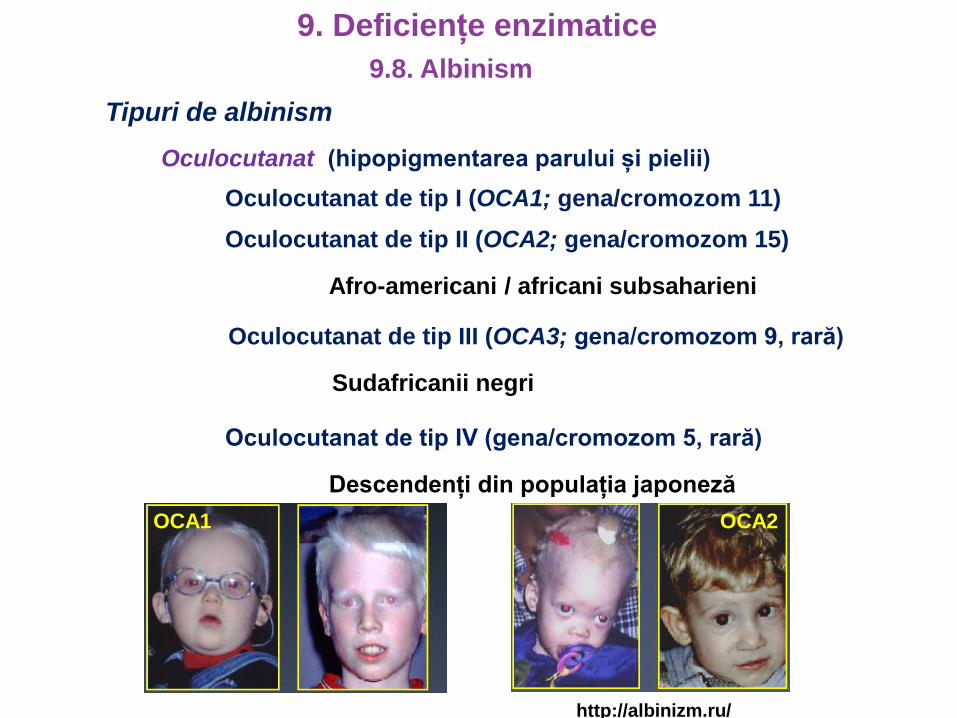
Tipuri de albinism
Oculocutanat (hipopigmentarea parului și pielii)
Oculocutanat de tip I (OCA1; gena/cromozom 11)
Oculocutanat de tip II (OCA2; gena/cromozom 15)
Afro-americani / africani subsaharieni
Oculocutanat de tip III (OCA3; gena/cromozom 9, rară)
Sudafricanii negri
Oculocutanat de tip IV (gena/cromozom 5, rară)
Descendenți din populația japoneză
OCA1 OCA2
http://albinizm.ru/
9.8. Albinism
9. Deficiențe enzimatice
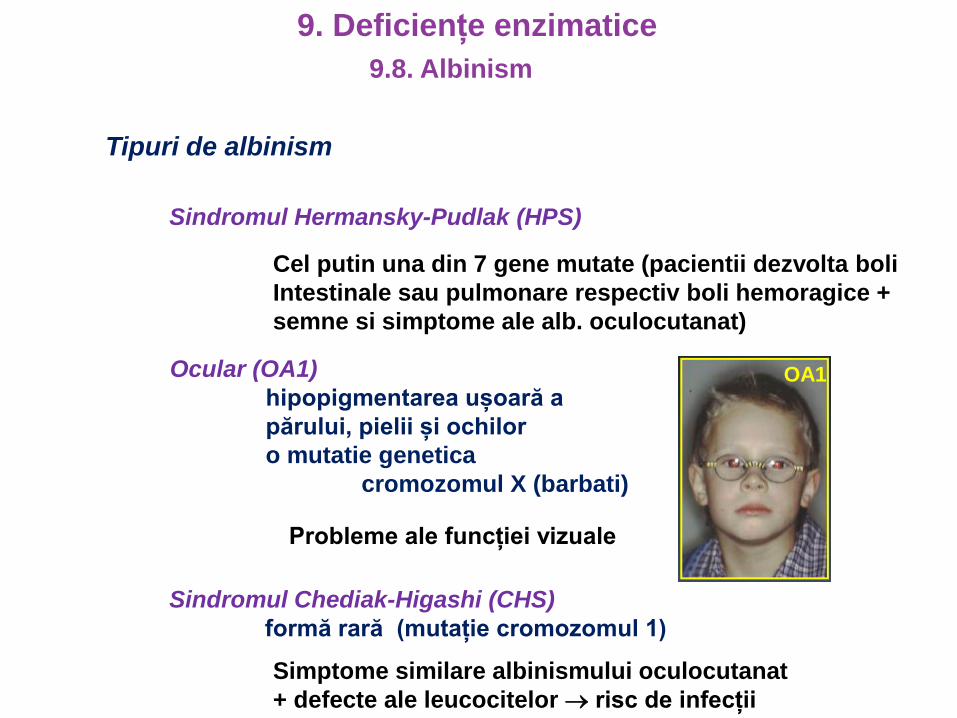
Tipuri de albinism
Ocular (OA1)
hipopigmentarea ușoară a
părului, pielii și ochilor
o mutatie genetica
cromozomul X (barbati)
Probleme ale funcției vizuale
Sindromul Chediak-Higashi (CHS)
formă rară (mutație cromozomul 1)
Simptome similare albinismului oculocutanat
+ defecte ale leucocitelor risc de infecții
Sindromul Hermansky-Pudlak (HPS)
Cel putin una din 7 gene mutate (pacientii dezvolta boli
Intestinale sau pulmonare respectiv boli hemoragice +
semne si simptome ale alb. oculocutanat)
OA1
9.8. Albinism
9. Deficiențe enzimatice
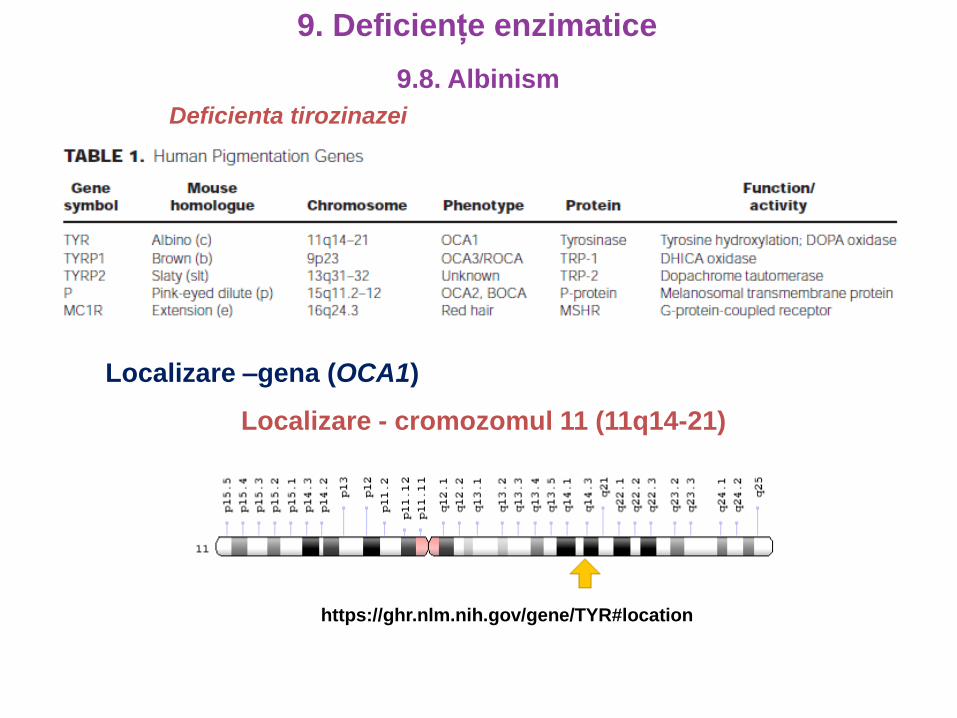
Localizare –gena (OCA1)
Localizare - cromozomul 11 (11q14-21)
https://ghr.nlm.nih.gov/gene/TYR#location
Deficienta tirozinazei
9.8. Albinism
9. Deficiențe enzimatice

Deficienta tirozinazei
Albinismul oculocutanat de tip I (OCA1)
Incidența 1 la 16.000-20.000
Boala autozomala recesivă
OCA 1 – 4 subtipuri
OCA 1A
OCA 1B
OCA 1MP
OCA 1TSOCA 1A – caracteristici:
Mutație nulă a genei tirozinazei
nu se produce melanina
Fenotip sever: păr alb, transiluminarea irisului, nistagmus
reducerea acuității vizuale (20/200-20/400)
Pacienții – risc pentru cancer
http://plasticsurgerykey.com/
9.8. Albinism
9. Deficiențe enzimatice
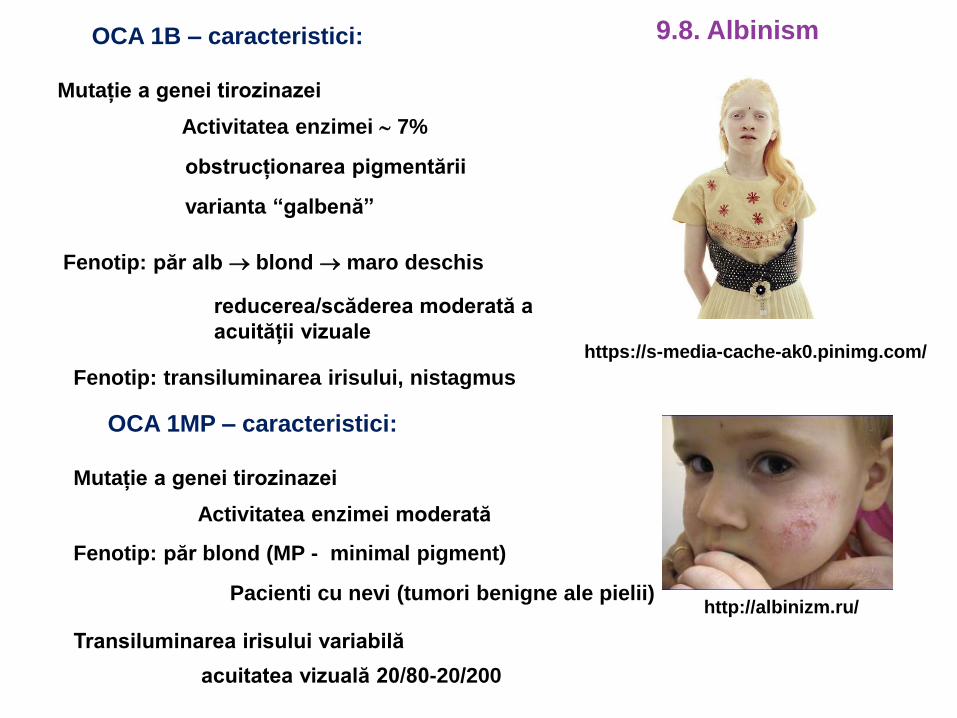
OCA 1B – caracteristici:
Mutație a genei tirozinazei
Activitatea enzimei 7%
Fenotip: păr alb blond maro deschis
reducerea/scăderea moderată a
acuității vizuale
obstrucționarea pigmentării
varianta “galbenă”
Fenotip: transiluminarea irisului, nistagmus
https://s-media-cache-ak0.pinimg.com/
OCA 1MP – caracteristici:
Mutație a genei tirozinazei
Activitatea enzimei moderată
Fenotip: păr blond (MP - minimal pigment)
Pacienti cu nevi (tumori benigne ale pielii)
Transiluminarea irisului variabilă
acuitatea vizuală 20/80-20/200
http://albinizm.ru/
9.8. Albinism

OCA 1TS – caracteristici:
Mutație a genei tirozinazei
Producerea unei enzimei - activitate f(T)
Fenotip: păr închis pe brațe, picioare și piept
Fenotip: păr alb (axilar, pubian, scalp)
La naștere nu poate fi diferențiat față de OCA1A
Tirozinaza- activitate normală în zonele mai “reci” ale organismului
Tirozinaza- activitate în zonele mai “calde” ale organismului
http://www.handresearch.com/
Levin, A.V. & Stroh, E., Journal of AAPOS,
15 (1), 59-66, 2011
9.8. Albinism
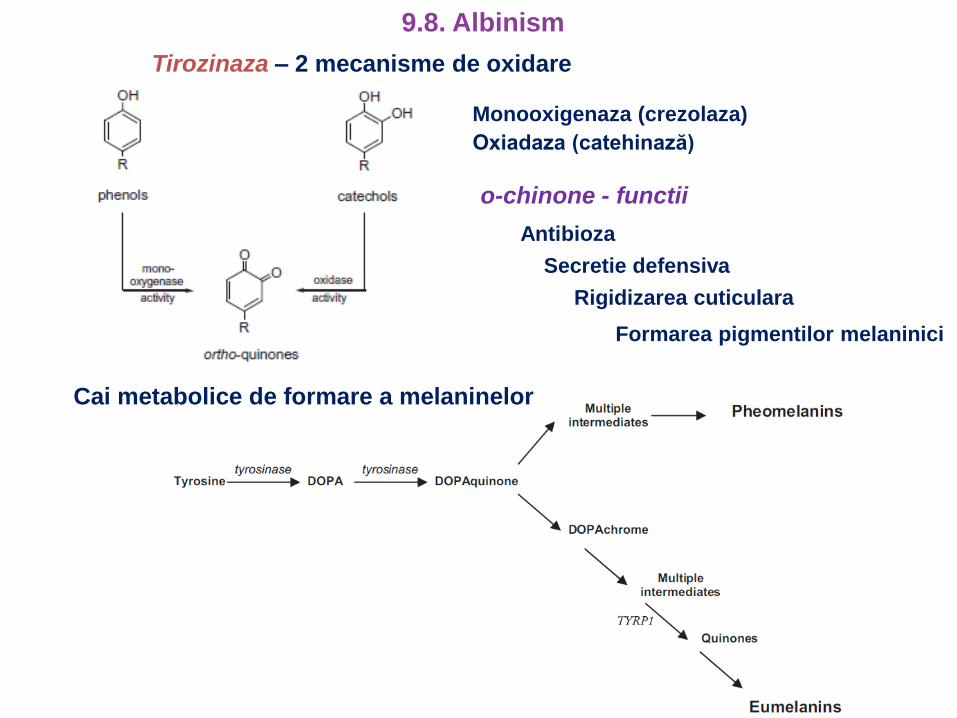
Cai metabolice de formare a melaninelor
Tirozinaza – 2 mecanisme de oxidare
o-chinone - functii
Antibioza
Secretie defensiva
Rigidizarea cuticulara
Formarea pigmentilor melaninici
Monooxigenaza (crezolaza)
Oxiadaza (catehinază)
9.8. Albinism
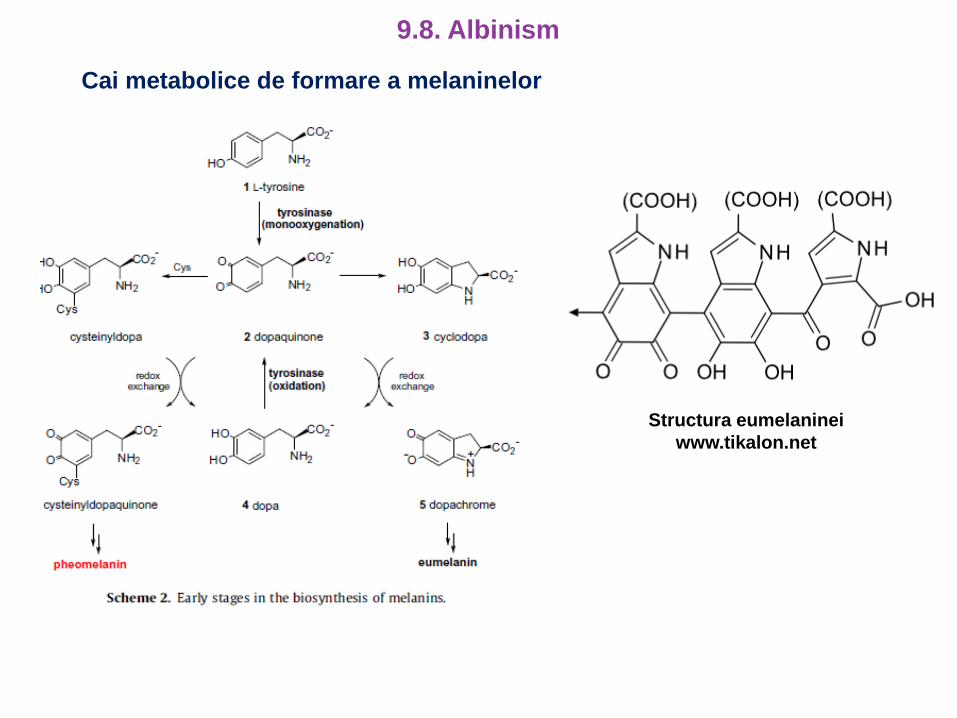
Cai metabolice de formare a melaninelor
Structura eumelaninei
www.tikalon.net
9.8. Albinism
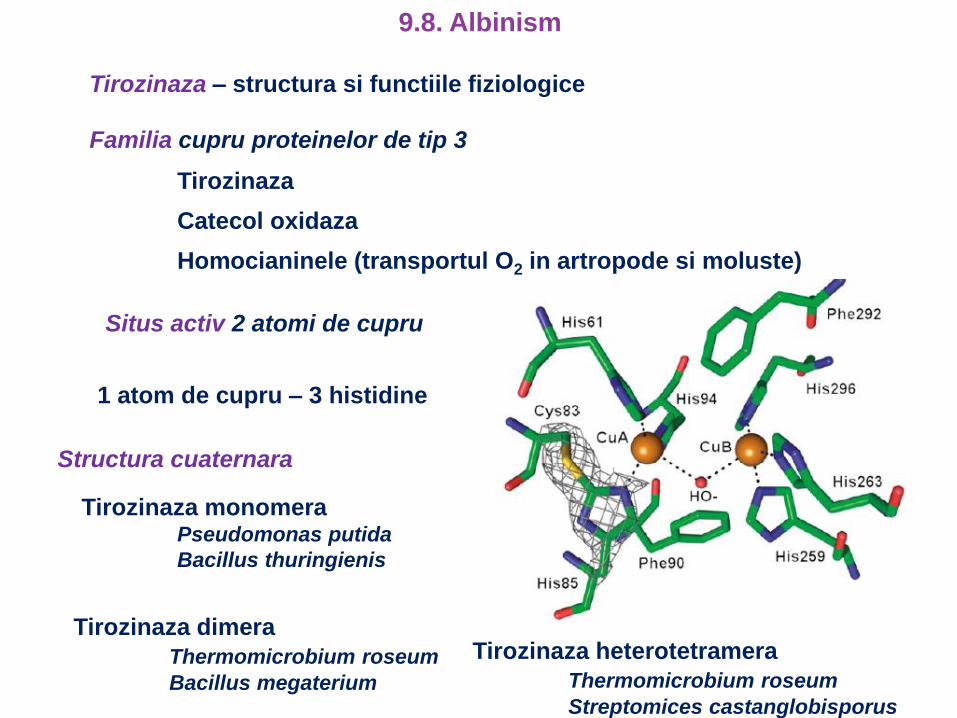
Tirozinaza – structura si functiile fiziologice
Familia cupru proteinelor de tip 3
Tirozinaza
Catecol oxidaza
Homocianinele (transportul O2 in artropode si moluste)
Situs activ 2 atomi de cupru
1 atom de cupru – 3 histidine
Structura cuaternara
Tirozinaza monomeraPseudomonas putida
Bacillus thuringienis
Tirozinaza dimera
Thermomicrobium roseum
Bacillus megaterium
Tirozinaza heterotetramera
Thermomicrobium roseum
Streptomices castanglobisporus
9.8. Albinism
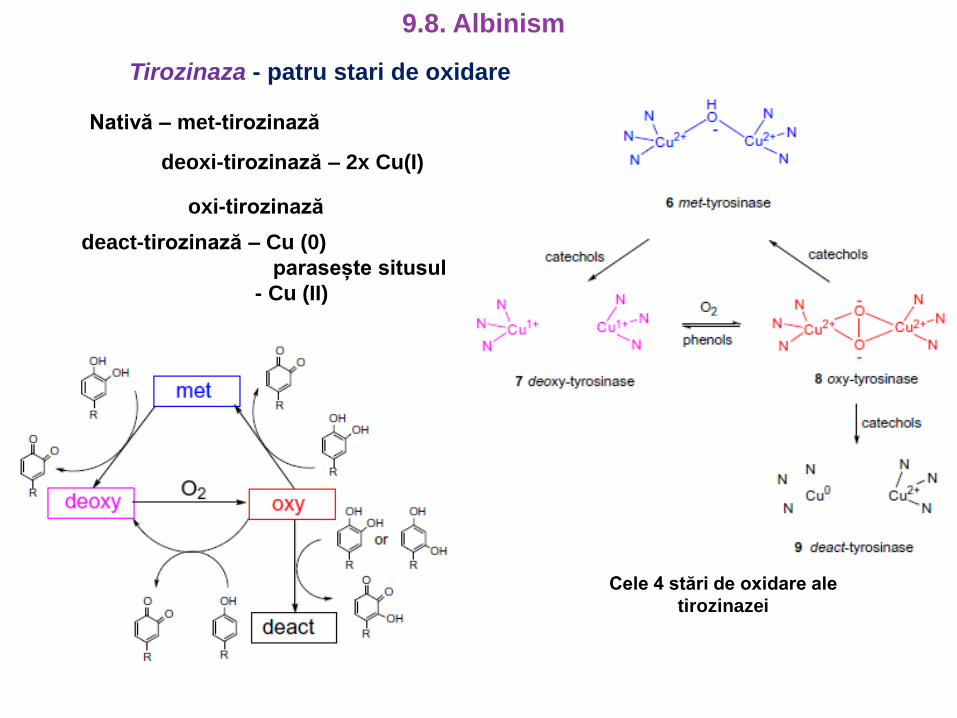
Tirozinaza - patru stari de oxidare
Nativă – met-tirozinază
deoxi-tirozinază – 2x Cu(I)
oxi-tirozinază
deact-tirozinază – Cu (0)
parasește situsul
- Cu (II)
Cele 4 stări de oxidare ale
tirozinazei
9.8. Albinism
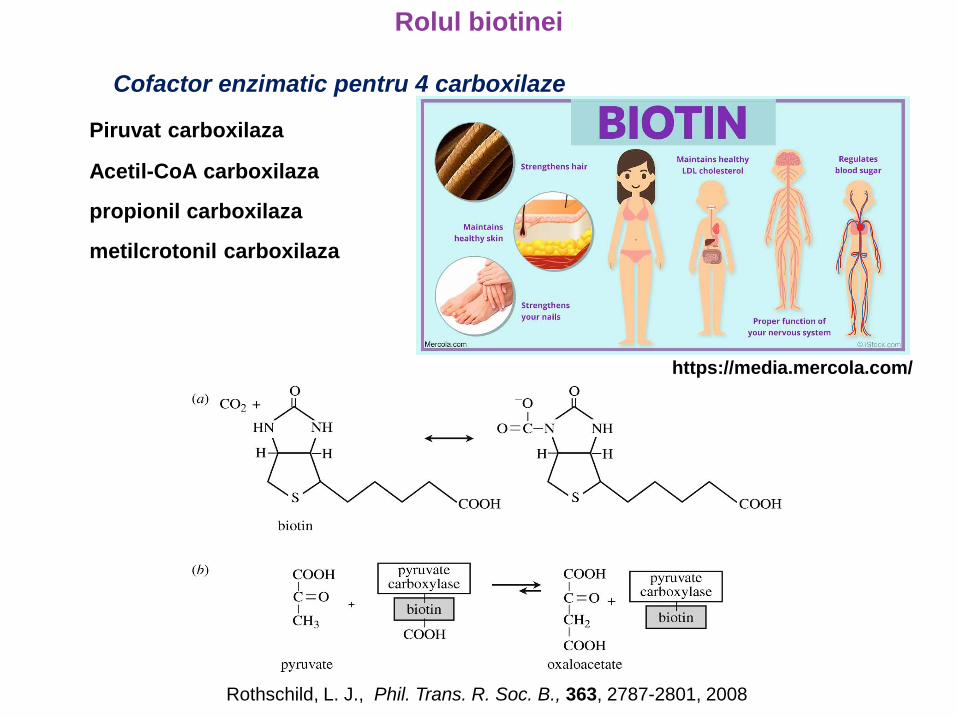
Rolul biotinei
Cofactor enzimatic pentru 4 carboxilaze
Piruvat carboxilaza
Acetil-CoA carboxilaza
propionil carboxilaza
metilcrotonil carboxilaza
Rothschild, L. J., Phil. Trans. R. Soc. B., 363, 2787-2801, 2008
https://media.mercola.com/
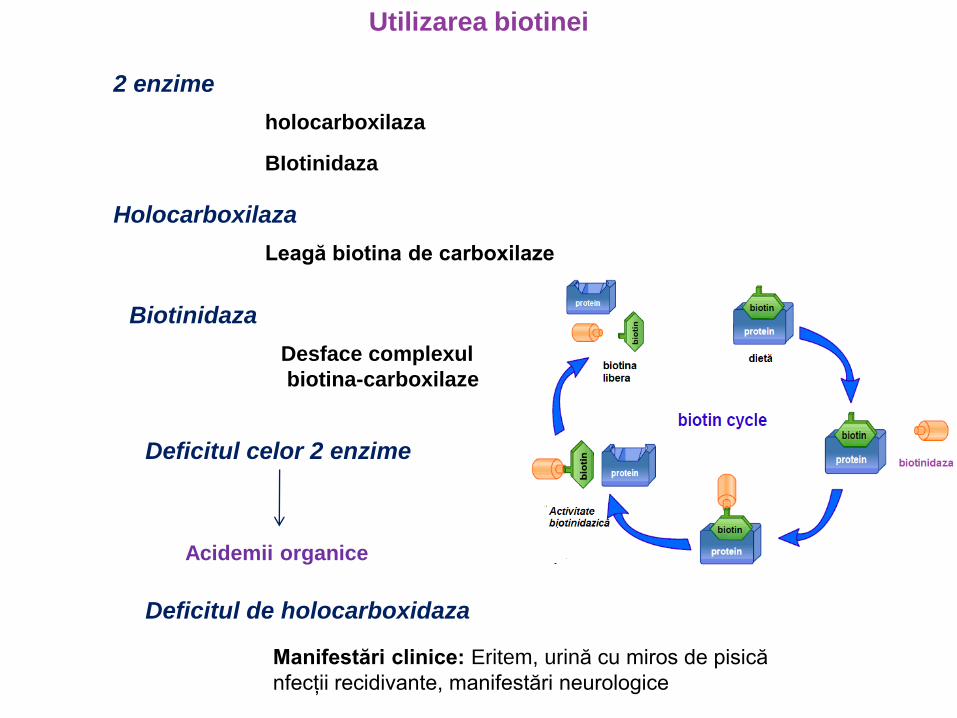
Utilizarea biotinei
2 enzime
holocarboxilaza
BIotinidaza
Holocarboxilaza
Leagă biotina de carboxilaze
Biotinidaza
Desface complexul
biotina-carboxilaze
Deficitul celor 2 enzime
Acidemii organice
Deficitul de holocarboxidaza
Manifestări clinice: Eritem, urină cu miros de pisică
nfecții recidivante, manifestări neurologice
Icterul
Localizare - 3p25.1
Deficienta biotinidazei
Incidența (1/60.000 SUA)
https://ghr.nlm.nih.gov/
Transmitere autozomala recesiva
Debut: sugar/copil mic
Manifestări clinice: cutanate (dermatite), convulsii, surditate, atrofie optică,
întârzieri în dezvoltare
Tratament: administrarea de biotină
Lungime genă – 23 kb (4 exoni)
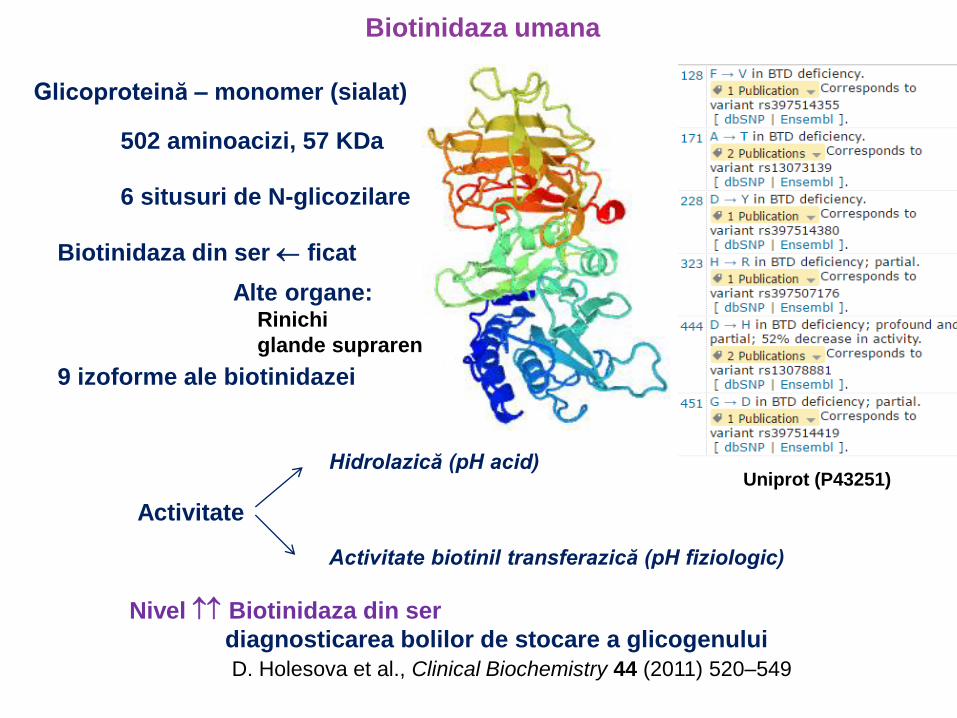
Biotinidaza umana
Glicoproteină – monomer (sialat)
502 aminoacizi, 57 KDa
6 situsuri de N-glicozilare
Biotinidaza din ser ficat
9 izoforme ale biotinidazei
Activitate
Hidrolazică (pH acid)
Activitate biotinil transferazică (pH fiziologic)
Alte organe: Rinichi
glande suprarenale
Nivel Biotinidaza din ser
diagnosticarea bolilor de stocare a glicogenului
D. Holesova et al., Clinical Biochemistry 44 (2011) 520–549
Uniprot (P43251)

vLCAD-2 (ACAD-9)
C16:0 ; cis-9-C16:1 ; cis-9,12-C18:2
Gel filtrareDimer
Ensenauer, R. et al.
JBC 90, Jun (2005)
vLCAD-1 / >C16
Gel filtrareDimer
Zhang, J. et al, BBRS
297, 1033-1042
ETF
ETF
DH
Lant respirator
i2VD (SBCAD) / iLeuCOSCoA
Uman+aceto-acetil-CoA
Tetramer, Res 2.6 Å
Preliminary data
i3VD / i3vCOSCoA
Tiffany, K. A. et al.
Biochemistry 36,
8455 (1997)
Tetramer, Res 2.6 Å
iBD / iBuCOSCoA
Uman + iBD-CoA
Battaile, K. P. et al.
JBC 279,
16526 (2004)
Tetramer, Res 1.8 Å
GD / GCOSCoA
Uman +4-NBCoA
Fu, Z et al.
Biochemistry 43,
9674 (2004)
Res 2.1 Å
e-
e-
2
e-
R3
|
R1— C=C—COSCoA
|
R2
+2H+
MCAD/C6—C10
S.s. +C8CoA
Kim, J.-J. et al.
PNAS 90, 7523
(1993)
Res 2.4 Å
SCAD/C4—C6
M.e. + 3-keto-C4CoA
Djordjevic, S et al.
Biochemistry 34,
2163 (1995)
Res 2.5 Å
LCAD/C10—C18; C12
Eder, M. et al, EJB,
245, 600-607
Gel filtrareTetramer
-oxidarea
mitochondriala
Catabolismul
aminoacizilor
Oxidarea acizilor grasi
Deficienta ACAMD
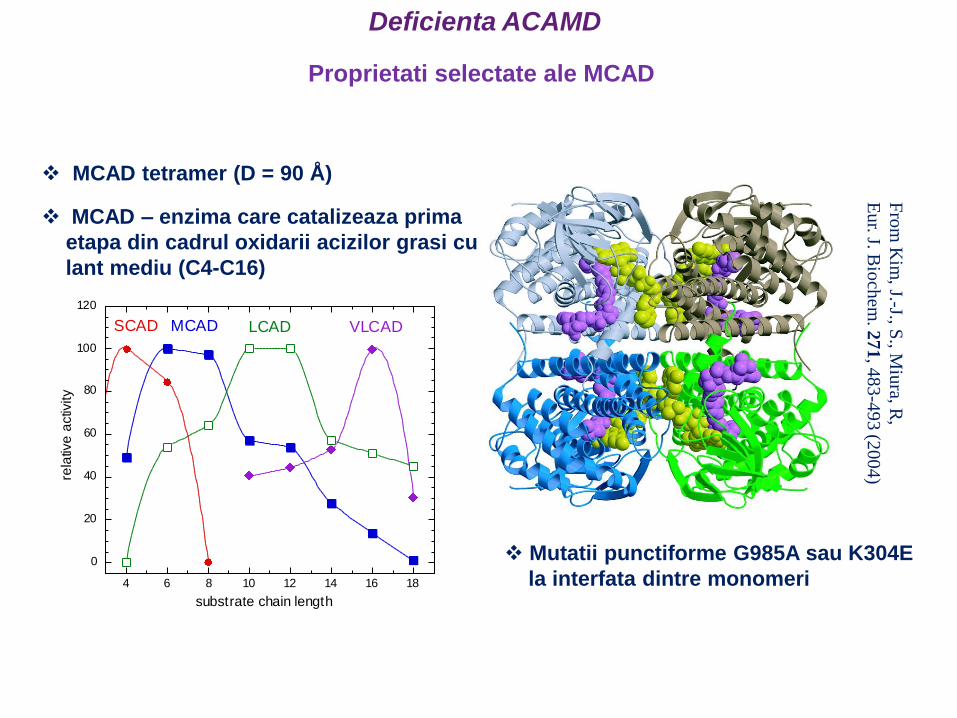
0
20
40
60
80
100
120
4 6 8 10 12 14 16 18
rela
tive
activ
ity
substrate chain length
LCAD VLCADMCADSCAD
MCAD tetramer (D = 90 Å)
MCAD – enzima care catalizeaza prima
etapa din cadrul oxidarii acizilor grasi cu
lant mediu (C4-C16)
Mutatii punctiforme G985A sau K304E
la interfata dintre monomeri
Fro
m K
im, J.-J., S
., Miu
ra, R,
Eu
r. J.B
ioch
em.2
71
, 48
3-4
93 (2
00
4)
Proprietati selectate ale MCAD
Deficienta ACAMD
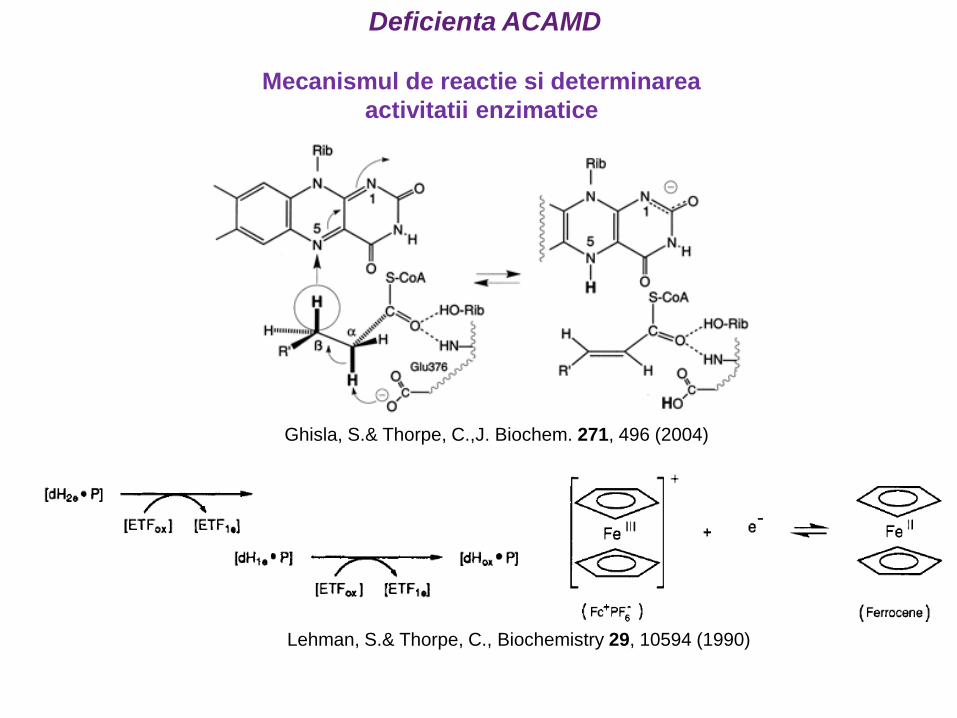
Ghisla, S.& Thorpe, C.,J. Biochem. 271, 496 (2004)
Lehman, S.& Thorpe, C., Biochemistry 29, 10594 (1990)
Mecanismul de reactie si determinarea
activitatii enzimatice
Deficienta ACAMD


















