







Limbile
Pagini
Legal


Aspecte privind proprietatile fizice si termodinamica fenomenologica
a solutiilor
INTRODUCERE
Propietãţile macroscopice şi de transport ale amestecurilor lichide sunt constituie o
reflactare a forţelor intermoleculare şi a structurii microscopice a lichidelor. Cunoaşterea unor
propietãţi fizice precum şi densitatea, indicele de refracţie, viscozitatea, constanta dielectricã
este esenţialã pentru înţelegerea interacţiunilor dintre diferitele molecule de soluţii prezentate
in soluţiile lichide.
Variaţia acestor propietãţi cu concentraţia furnizeazã informaţii importante despre
interacţiunile intermoleculare şi structura solventului.
Pentru înţelegerea comportamentului fizico-chimic al amestecurilor binare de alcanoli
cu MTBE au fost effectuate în ultimii ani cercetãri sistematice referitoare la propietãţile
termodinamice ale acestor sisteme.
Scopul şi conţinutul lucrãrii
Pentru a obţine date noi cu privire la natura soluţiilor constituite din metal-terţ-butil-
eter şi 1-propanol s-au studiat unele propietãţi fizico-chimice ale acestor sisteme. Studiul se
referã la determinãrile experimentale de viscozitate şi de constantã dielectricã precum şi
calculul şi discuţia unor mãrimi derivatedin aceste propietãţi precum: viscozitatea de exces,
mãrimi de activare caracteristice ale procesului de curgere, polarizare molecularã si
polarizare molecularã de exces.
Lucrarea este structuratã în douã parţi:-- în parte întâi este prezentat un studiu de
literaturã referitor la unele propietãţi fizice ale lichidelor, totodatã sunt prezentate succint
teorii ale stãrii lichidelor care şi-au adus un important aport în dezvoltarea domeniului
analizat. Astfel:
PARTEA I
Pagina 1 din 82

În capitolul I sunt prezentate câteva propietãţi fizice din care enumãr: viscozitatea
(generalitãţi, influenţa temperaturii asupra viscozitãţii, viscozitatea amestecurilor binare) şi
constanta dielectricã.
Capitolul II cuprinde tratarea din punct de vedere termodinamic, fenomenologic a
soluţiilor şi teorii de reţea, modelul potenţialului mediu şi teoria structurilor semnificative ale
lichidelor.
PARTEA A-II –A
În aceastã parte a lucrãrii conţine contributţii originale la caracterizarea unor soluţii de
metil-terţ-butil-eter si 1-propanol.
Capitolul III conţine valorile experimentale ale mãsurãtorilor de viscozitate ale
componenţilor puri si ale soluţiilor analizate pentru fracţii molare ale propanolului de:
0,1517; 0,3040; 0,4094; 0,5002; 0,6095; 07575; 0,9099; la temperaturile de 200 C, 250C, 300C
şi 350C. Se prezintã şi se discutã variaţia viscozitãţii cu temperatura şi concentraţia şisunt
prezentate metode experimentale utilizate pentru determinarea viscozitãţii lichidelor studiate.
În capitolul IV se comparã valorile viscozitãţii soluţiilor determinate expetimental cu
valorile calculate cu ajutorul a diverse ecuaţii propuse în literatura de specialitate pentru
calculul viscozitãtii soluţiilor binare lichide: viscozitatea idealã Arrhenius, ecuaţiile
Dolezalek, Grumberg-Nissan si Vijk.
Au fost calculate şi discutate de aemenea valorile viscozitãţii de exces şi este
reprezentatã variaţia acestei mãrimi de exces cu concentraţia şi cu temperatura. Capitolul
conţine valorile coeficienţilor din ecuaţia polinimialã Redlich-Kister care coreleazã valorile
viscozitãţii de exces ale soluţiilor.
Sunt prezentate de asemena mãrimile specifice de activare ale procesului de curgere;
energia de activare şi factorul preexponenţial.
Capitolul V conţine rezultaeale experimentale ale mãsurãtorilor de constantã
dielectricã efectaute pentru componenţii puri şi pentru soluţiile de metil-terţ-butil-eter şi 1-
propanol la temperaturile de 200 C, 250C, 300C şi 350C. Capitolul mai conţine şi valorile
polarizãrii moleculare şi ale polarizãrii molecualre de exces ale soluţiilor de metil-terţ-butil-
eter şi 1-propanol.
Este ilustratã variaţia polarizãrii molecualre de exces în funcţie de concentraţia
soluţiei exprimatã în funcţii molare la temperaturile menţionate mai sus.
Pagina 2 din 82

În capitolul VI sunt prezentate concluziile lucrãrii elaborate.
Capitolul VII conţine unele aspecte metodologice şi un proiect de lecţie pentru clasa a
–XII-a având ca temã ”Alcooli, generalitãţi, metode de obţinere”.
Capitolul I
PROPRIETATI FIZICE ALE LICHIDELOR
I.1 VISCOZITATEA LICHIDELOR
I.1.1 Generalitati
Fenomene de transport in lichide
Dintre fenomenele de transport in lichide, de o deosebitã atenţie s-a bucurat
viscozitatea, pentru a cãrei mãsurare au fost dezvoltate tehnici experimentale exacte şi
aparate ingenioase. Bogãţia si precizia datelor experimentale acumulate au stimulat
elaborarea a numeroase teorii privind viscozitatea lichidelor. De aceea, dintre fenomenele de
transport în lichide, viscozitatea formeazã principalul conţinut al acestui capitol.
Ca şi în gaze, forţa care solicitã un strat de lichid adiacent, este proporţionalã cu
suprafaţa acesteia şi cu gradientul de vitezã , normal pe direcţia de mişcare. Acest enunţ
constituie legea lui Newton privitoare la viscozitate. Pentru unitatea de suprafaţã, forţa de
frecare f este datã de ecuatia:
(1)
în care factorul de proporţionalitate η reprezintã coeficientul de viscozitate.
Valorea reciprocã φ a viscozitaţii este numitã fluiditate.
(2)
iar raportul dintre viscozitate şi densitatea lichidului se numeşte viscozitate
cinematicã.
Spre deosebire de coeficienţii de viscozitate ai gazelor, coeficienţii de viscozitate ai
lichidelor scad sensibil când temperature lichidelor creşte. De aceea, valabilitatea ecuaţiilor şi
Pagina 3 din 82

teoriilor viscozitaţii lichidelor se apreciazã dupã exactitatea cu care redau variaţia viscozitaţii
cu temperatura.
Cea mai mare parte din ecuaţiile viscozitaţii lichidelor sunt rezultatul nemijlocit al
unor teorii ale viscozitaţii şi vor fi prezentate în legãturã cu aceste teorii. Sunt insã şi câteva
ecuaţii empirice, de realã unitate, pe care urmeazã sã le considerãm mai întãi.
Ecuaţii empirice ale viscozitãţii lichidelor
Pentru a reda variaţia coeficientului de viscozitate cu temperatura absolutã, Arrhenius
şi Guzman au propus, independent unul de celãlalt ecuaţia polinomialã:
(3)
în care A şi B sunt constante, ultima având dimensiunile unei energii.
Aşa cum rezultã din legea lui Newton coeficientul de viscozitate are dimensiunea
şi şi unitate de mãsurã în sistemul C.G.S. numitã poisse, reprezintã .
Fiind prea mare, se preferã sã se exprime coeficienţii de viscozitate în centi sau milipoisse.
Ecuaţia (3) nu se aplicã in cazul lichidelor polare. La unele lichide, chiar nepolare,
ecuaţia nu redã cu fidelitate datele experimentale. Totuşi ea a fost aplicatã cu succes la
numeroase lichide atât moleculare cât şi ionice dar şi metalice.
O altã ecuaţie a viscozitãţii, caracterizatã printr-o remarcabilã simplitate, este ecuaţia
lui Batschiscki:
(4)
În care v este volumul specific, iar c şi ω sunt douã constante care depind de natura
lichidului. Semnificaţia constantei ω, numitã volumul specific limitã, rezultã imediat; când v
tinde catre ω, viscozitatea devine infinitã, adicã lichidul se solidificã.
Volumul specific la limitã reprezintã deci volumul pãrţii rigide din lichid, iar diferenţa
v-ω ar fi volumul liber, disponibil pentru curgerea viscoasa. Constanta c are dimensiunile:
viscozitate∙volum.
Înmulţind numãrãtorul şi numitorul ecuaţiei (4) cu masa molecularã se obţine ecuaţia
lui Batschiscki, exprimatã in marimile moleculare corespunzãtoare:
(5)
pe care o vom prefera adesea în cele ce urmeazã.
Pagina 4 din 82

De observat cã ecuaţia lui Batschiscki redã explicit variaţia coeficientului de
viscozitate cu temperatura ci numai cu volumul sub forma implicitã a variaţiei volumului
specific v sau a volumului molar V.
Scriind ecuaţia sub forma:
(6)
şi punând, într-un sistem de douã axe rectangulare, fluiditatea pe abscise si volumul
specific pe ordonate, trebuie sã se obţinã pentru diferite lichide, linii drepte ale cãror
coeficieţi unghiulari dau constante c, iar ordonatele lor origine , volume specifice limite, ω.
Aceastã cerinţã a fost verificatã de Batschiscki la 87 de lichide moleculare ale cãror
viscozitãţi fuseserã determinate experimental de Thorpe şi Rodger. Cu puţine şi
nesemnificative abateri, ecuaţia sa s-a devenit perfect valabilã.
I.1.2 Influenţa temperaturii asupra viscozitãţii
În toate cazurile viscozitatea lichidelor scade foarte repede odatã cu ridicarea
temperaturii, respectiv creşte foarte repede dacã temperatura scade. Aceastã variaţie este în
principiu exponenţialã pentru lichidele cu o curgere obişnuitã, adicã variaţia viscozitãţii
(7)
Comparând aceastã expresie se poate vedea cã U este energia de activare , raportatã la
1 mol, a mobilitãţii moleculare de lichid, iar factorul A dinaintea exponenţialei nu poate fi în
mod riguros independent de temperaturã. Aceastã dependenţã este însã neglijabilã în raport
cu rapiditatea variaţiei exponenţialei. Din demonstraţiile de mai sus rezutã şi faptul cã energia
de activare U, este caracteristicã viscozitãţii şi difuziunii , este aceeaşi. Experienţa verificã
aceastã concluzie.
Variaţia exponenţialã simplã cu temperatura, aşa cum rezultã din ecuaţia (7) este
valabilã însã în numeroase cazuri numai într-un interval de temperaturi nu prea mare, adicã
diagrama lg cuprinde adeseori porţiuni mai mari în care
reprezentarea nu este o dreaptã. Acest fapt se poate datora mai multor cauze. Astfel, în cazul
lichidelor care nu sunt chiar simple, odatã cu creşterea temperaturii se poate modifica stuctura
lor (de exemplu se pot rupe succesiv punţile de hidrogen), prin urmare nici numãrul golurilor
care determinã curgerea nu mai creşte dupã o simplã relaţie exponenţialã.
Pagina 5 din 82

Însã în acelaşi timp se poate modifica şi mãrimea medie a golurilor, şi datoritã
structurii modificate a mediului se schimbã şi energia de activare a salturilor. În cazul unor
molecule mari, de formã aproape simetricã, la temperaturi mai joase ordinea este relativ mai
mare şi din cauza aşezãrii reciproce paralele, golurile sunt mici; dar, odatã cu creşterea
temperaturii se mãreşte dezordinea şi, deoarece la moleculele mari aşezarea reciprocã
paralelã se face mai greu, cresc nu numai numãrul, ci şi dimensiunile golurilor, micşornâdu-
se mult vicozitatea.
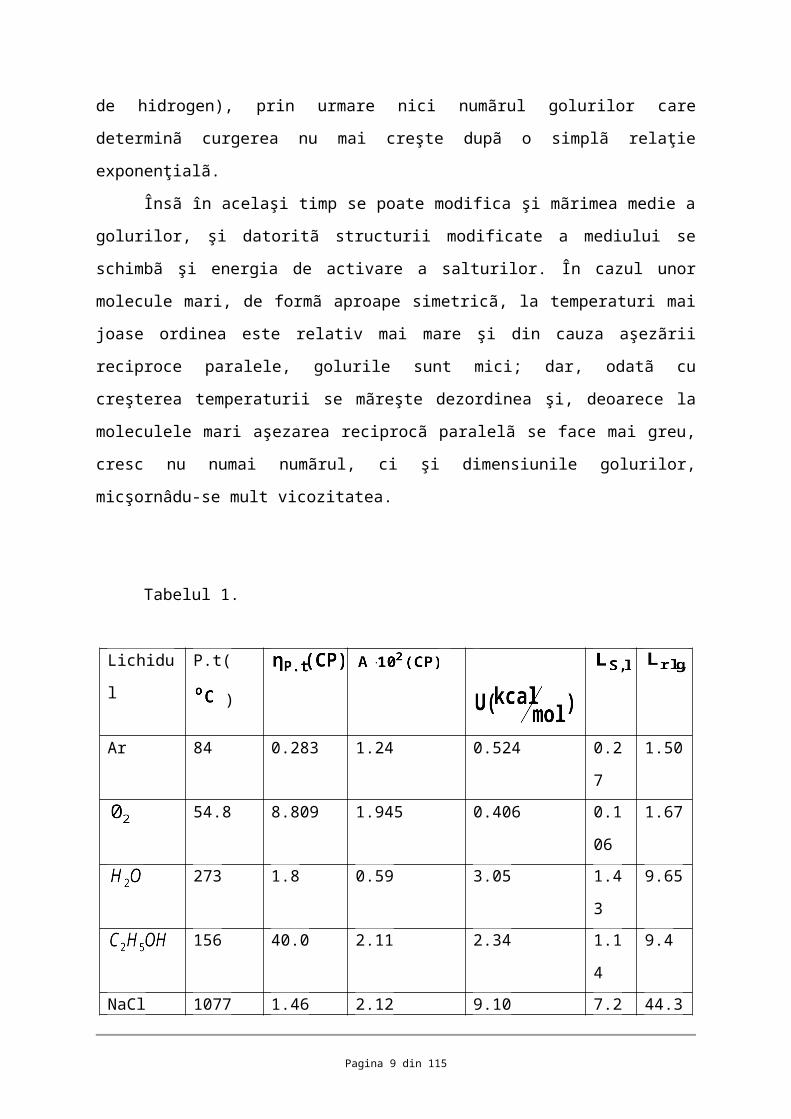
Tabelul 1.
Lichidul P.t( )
Ar 84 0.283 1.24 0.524 0.27 1.50
54.8 8.809 1.945 0.406 0.106 1.67
273 1.8 0.59 3.05 1.43 9.65
156 40.0 2.11 2.34 1.14 9.4
NaCl 1077 1.46 2.12 9.10 7.22 44.3
Ag.. 1233 4.14 56.9 4.87 2.63 59.5
Hg... 234 2.01 55.5 0.598 0.57 14.0
În tabelul 1 sunt date viscozitãţile unor lichide la punctul lor de topire, precum şi
valorile caracteristice A şi U cu ajutorul cãrora se poate calcula dupã ecuaţia (7) variaţia
viscozitãţii în apropierea punctului de topire. Energiile de activare U pot fi comparate cu
valorile cãldurilor de topire şi vaporizare specificate de asemenea în tabel. Se poate vedea cã
U este de fapt mai mare decât caldura de topire, dar nu o depãşeşte cu mult. Deoarece şi
topirea a fost întreruptã cu ajutorul mecanismului prin goluri aceastã concordanţã
aproximativã este o nouã dovadã în favoarea mecanismului prin goluri al frecãrii interne.
Datele cuprinse în tabelul 1 permit încã o serie de comparaţii interesante. De exemplu,
comparând energia de activare a bromurii de etil cu aceea a alcoolului etilic, se poate vedea
rolul puntilor de hidrogen în viscozitate: la aproape aceeaşi temperaturã viscozitatea
alcoolului este mult mai mare. Dupã cum se ştie, aceastã acţiune de mãrire a viscozitaţii
Pagina 6 din 82

exercitatã de punţile de hidrogen se manifestã mult mai puternic la polialcooli (glicoli,
glicerinã).
Comparând însã alcoolul cu apa, care în apropierea punctului de topire se gaseşte de
asemenea cã valoarea constantei A pentu apã este foarte mica. Acest fapt poate fi explicat cu
ajutorul mãrimilor golurilor; ori, se ştie cã structura apei în apropierea punctului de topire
este reticularã şi foarte afânatã, ceea ce concordã foarte bine cu aceastã constatare.
În ceea ce priveşte factorul A, un caz interesant îl constituie metalele: faptul cã
golurile sunt foarte mici, ceea ce este în concordanţã cu faptul cã metalele topite, ca şi
cristalele metalelor, nu sunt formate din atomi metalici liberi, ci din ionii acestora care au
dimensiunile mult mai mici.
Aceastã concluzie rezultã şi din faptul cã metalele topite prezintã coductibilitatea
electronica ca şi cristalele lor.
Rolul hotãrâtor al golurilor în frecarea internã mai rezultã şi dintr-o relaţie empiricã
stabilitã de Batschiscki ( 1913 ), şi anume:
(8)
în care :
C - constanta caraceristicã diferenţelor lichide
V-volumul real al lichidelor
- volumul minim la care poate fi comprimat lichidul
Temperatura nu intervine în modul explicit în ecuaţia lui Batschiscki, dar ecuaţia
cuprinde totuşi variaţia lui η cu temperatura (deci viscozitatea, în concordanţã cu experienţa,
scade). din numitorul ecuaţiei nu reprezintã altceva decât volumul "liber" al
lichidului, cu alte cuvinte volumul golurilor.
Dacã se considerã cã la o presiune datã volumul unui gol este constant atunci
creşterea volumului liber odatã cu creşterea temperaturii trebuie atribuitã mãrimii lichidelor,
ca şi al cristalelor dupã o lege exponenţialã, aşa cã în fond formula lui Batschiscki şi ecuaţia
(7) exprima acelaşi lucruri.
I.1.3 Viscozitatea amestecurilor binare lichide
Viscozitatea este o proprietate de transport care dã indicaţii asupra frecãrii interne a
lichidelor şi prezintã importanţã în legãturã cu proiectarea şi operarea instituţilor industriale.
Pagina 7 din 82

La lichide viscozitatea scade cu creşterea temperaturii şi creşte odatã cu creştere
presiunii. Variaţia viscozitãţii cu temperatura exprimatã de o relaţie exponenţialã prezentatã
de Guzman şi Carrancio în 1913:
(9)
în care este factorul preexponenţial iar este energia de activare a curgerii.
Relaţia este o relaţie liniarã datoritã compensãrii variaţiei cu
temperatura factorului exponenţial şi a energiei de activare a curgerii.
Viscozitatea solutiilor binare lichide
Viscozitatea amestecurilor binare lichide este un subiect de interes general. În prezent
nici o teorie statisticã nu poate fi utilizatã cu succes pentru calculul viscozitãţii amestecurilor
binare, astfel încât viscozitatea se exprimã prin legi empirice de forma:
(10)
în care x şi (1-x) sunt fracţiile molare iar şi viscozitaţile componenţilor puri.
Totuşi viscozitãţile majoritãţii sistemelor binare nu pot fi reprezentate adecvat prin
ecuaţia de mai sus, când f(η) este η însãşi, log η, sau . Hind şi McLaughlin utilizeazã o
ecuaţie de forma:
(11)
unde este un parametru de interacţiune.
În ultimele decenii, au fost efectuate multe studii în vederea prezicerii teoretice a
acestei proprietãţi de transport. Au fost definite un numãr de diferite relaţii (empirice sau
semiempirice) reprezentând modul în care aceastã mãrime variazã cu concentraţia, în care
unul sau câţiva parametri adjustabili permit stabilitatea abaterii de la idealitate. În funcţie de
autori, idealitatea este caracterizatã printr-o relaţie liniarã între concentraţie şi viscozitate cu
logaritmul viscozitãţii.
Înainte de aplicarea acestor ecuaţii este necesar sã se dispunã de valori experimentale
ale viscozitãţii soluţiei în scopul determinãrii parametrilor adjustabili. Aceastã expresie
coreleazã viscozitatea soluţiei binare cu proprietãţile termodinamice ale celor douã reacţii
absolute şi pe teoria vitezei de reacţie absolute, care este asociatã cu energia liberã de
activare. În acest fel, autorii evalueazã viscozitatea de exces a amestecurilor lichide
presupunând cã energia liberã de exces a amestecului este egalã cu energia liberã de exces a
Pagina 8 din 82
activãrii. Pentru aceastã abordare, toate mãrimile termodinamice implicate în ecuaţie sunt
calculate prin teoria Statisticã a soluţiilor elaboratã de Flory. Aceastã relaţie a fost utilizatã cu
succes şi de alţi cercetãtori pentru a descrie viscozitatea diverselor amestecuri binare.
Pentru a calcula viscozitatea amestecurilor lichide Grumberg şi Nissan au propus o
ecuaţie empiricã de calcul:
(12)
unde parametrul d reflectã neidealitatea sistemului. Acest parametru este o mãsurã
aproximativã a teoriei interacţiunii dintre componenţi.
Totuşi s-a arãtat cã în anumite cazuri el poate fi de asemenea corelat cu diferenţa
volumelor moleculare ale omponenţilor şi cu entropia de amestec.
Ecuaţia McAllister se bazeazã pe modelul propus de Eyring, care considerã cã interacţiunea are loc între trei particule:
în care ν reperezintã viscozitatea cinematicã iar şi sunt parametri de
interacţiune obişnuiţi prin metoda celor mai mici pãtrate.
Teja şi Rice au propus o metodã de calcul a viscozitãţilor soluţiilor lichide bazatã pe
metoda stãrilor crespondente:
14)
unde 1M şi 2M se referã la proprietãţile a douã fluide de referinţã (comparanţi
puri) şi este factorul acentric. Valorile se calculeazã cu formula:
(15)
în care şi sunt volumul critic si respectiv temperatura
criticã.
Comform modelului Schrodt şi Akel, bazat pe conceptul viscozitãţii fluidului a lui
Eyring, ecuaţia de calcul a viicozitãţii soluţiilor este:
Pagina 9 din 82

(16)
în care:
este coeficientul de activitate al viscozitãţii, iar este numãrul lui Avogadro.
Ecuaţia Auslander utilizatã şi de alţi autori permite calculul viscozitãţii soluţiilor pe
baza unor parametri adjustabili reprezentând interacţiuni binare:
(17)
Relaţiile de calcul a viscozitãţii amestecurilor binare lichide propuse de Dolezalek,
Grunberg-Nissan şi Wijik sunt prezentate în capitolul 4.
I.2. CONSTANTA DIELECTRICĂ
Dacã se plaseazã o substanţã într-un câmp elctric omogen putem distinge douã cazuri:
a) Dacã substanţa conţine purtãtori de sarcinã liberi (electroni, ioni), aceştia se vor
deplasa sub influenţa câmpului electric producându-se transportul de sarcinã;
b) Dacã substanţa nu conţine purtãtori de sarcinã liberã (substanţe neconducãtoare) ea
este un dielectric şi nu se produce transport de sarcinã. Prezenţa dielectricului micşoreazã
intensitatea câmpului electric.
Dacã şi sunt intensitãţile corpului electric în vid şi respectiv în prezenţa
dielectricului, rapotul: sau , se numeşte constantã
dielectricã a substanţei respective. Constanta dielectricã se poate defini şi cu relaţia: ,
unde şi reprezintã forţa care se exercitã între douã sarcini electrice în vid, respectiv
forţa care se exercitã între sarcinile electrice când între ele se plaseazã un dielectric.
Micşorarea intensitãţii corpului electric şi a forţei electrice într-un dielectric se poate
explica prin fenomenul de polarizare dielectricã a moleculelor.
Pagina 10 din 82

Polarizarea apare datoritã câmpului electric care se execitã asupra sarcinilor pozitive
şi a celor negative din molecule (atomi), forţe de sens opus, nucleul atomic şi electronic
deplasându-se în direcţii opuse, astfel încât centrul de greutate al sarcinilor pozitive se
depãrteazã de cel al sarcinilor negative: moleculele, respectiv atomii devin dipoli. Mãrimea
dipolului se caracterizeazã prin dipol momentul, care este produsul dintre mãrimea l a
sarcinilor aflate în centrul de greutate al sarcinilor pozitive şi acelor negative, şi distanţa d
dintre aceste douã centre de greutate.
Figura 1.2.
În cazul moleculelor polare (cu dipol permanent), centrele de greutate ale sarcinilor
pozitive şi negative nu coincid chiar şi în absenţa unui câmp electric. Prezenţa unui câmp
electric omogen mãreşte prin inducţie momentul electric al moleculelor polare. Aceastã
creştere a momentului electric formează momentul electric indus. Mărimea dipol-momentului
în absenţa câmpului depinde de structura moleculei, deci studiul momentului indus şi a celui
permanent poate servi la lămurirea unor probleme de structutã d a moleculei. Câmpul electric
Pagina 11 din 82

acţioneazã în mai multe feluri asupra moleculelor; în consecinţã şi polarizarea dielectricã se
compune din mai multe pãrţi:
1. Polarizarea electricã: învelişul electric al atomului respectiv al moleculei se
deplseazã în raport cu nucleul atomic, fãrã ca în interiorul moleculei poziţia relativã a
nucleelor atomice sã se modifice.
2. Polarizarea ionicã sau atomicã: dacã molecula (respectiv reţeaua cristalinã a
dielectricului) este formatã din ioni sau are o structurã polarã, atunci sub influenţa câmpului
electric, în interiorul moleculei (repectiv în reţeaua cristalinã) atomii şi ionii (împreunã cu
nucleele atomice respective) se deplaseazã în raport cu ceilalţi;
3. Polarizarea de orientare: dacã molecula are caracter polar în absenţa câmpului
exterior, atunci prezenţa câmpului orientarea dipolilor. Un câmp electric exterior tinde sã
orienteze paralel dipolii, ceea ce se întâmplã mai pronunţat cât intensitatea câmpului este mai
mare şi cu cât temperatura este mai joasã.
Polarizarea de orientare şi ionicã nu sunt independente una de cealaltã, deoarece odatã
cu orientarea dipolilor se deplaseazã şi nucleele atomice unele faţã de altele. Polarizarea
electronicã şi cea ionicã formeazã polarizarea de deplasare care depinde de temperaturã, pe
când polarizarea de orientare este invers proporţionalã cu intensitatea câmpului E:
(18)
Constanta de proporţionalitate este numitã polarizabilitatea (deformabilitatea)
moleculei.
Polarizabilitatea este dipol - momentul indus în molecula de dielectric de un câmp
cu o intensitate egalã cu unitatea. Ea aratã capacitatea de deformare a nucleelor atomice şi a
învelişului electronic fiind caracteristicã structurii moleculare. Polarizabilitatea este în
majoritatea cazurilor dependentã de direcţie (anizotropia polarizabilitãţii), de aceea în calcule
se lucreazã cu valorea sa medie . Numai în cazul moleculelor cu simetrie înaltã nu
depinde de direcţie.
I.1.2 Relaţia de polarizare dielectricã-constantã dielectricã
Ecuaţia Clausius-Mossotti
Pagina 12 din 82

Fie un condensator cu distanţa dintre armãturi d şi cu densitatea de sarcinã a
armãturilor (sarcina pe unitatea de suprafaţã: ) + e şi respectiv - e,conform figurii 1.3
- - - - - - - - - - - -
-
+ + + + + + + + + + + +
Figura 1.3
Intensitatea câmpului electric omogen în vid este:
Dacã între armãturi este un dielectric, atunci intensitatea câmpului scade:
Pentru un dielectric nepolar, acţiunea câmpului electric transformã molecula în dipoli
care îşi îndreaptã polii pozitivi spre armãtura negativã. În interiorul dielectricului, acţiunea
polilor pozitivi şi negativi induşi ai moleculelor se neutralizezã şi spre exterior acţioneazã
Pagina 13 din 82

dipolii de la suprafaţa dielectricului astfel încât neutralizeazã parţial acţiunea sarcinilor de pe
armãturi. Daca densitatea de sarcinã indusã este , intensitatea câmpului electric va fi:
(19)
Densitatea de sarcinã se numeşte polarizare şi este egalã cu: ; unde:
este numãrul de molecule din unitatea de volum ;
este momentul indus.
Rezultã:
(20)
Pentru calculul polarizãrii trebuie sã se cunoascã intensitatea câmpului care
acţioneazã asupra fiecãrei molecule în parte. Aceastã intensitate nu este egalã cu valoarea
calculatã cu relaţia (20) -- care exprimã intensitatea câmpului care acţioneazã asupra sarcinii
unor corpuri de dimensiuni macroscopice aflate într-un mediu continuu.
Dielectricul nu poate fi considerat ca un mediu continuu, ci trebuie sã se ţinã seama de
faptul cã moleculele se gãsesc în vidul aflat între celelalte molecule. Din aceastã cauzã,
intensitatea câmpului care acţioneazã asupra diferenţelor moleculare va fi de fapt mai mare
decât în cazul în care moleculele s-ar gãsi într-un mediu continuu cu constanta dielectricã ;
uneori, din cauza moleculelor vecine, intensitatea câmpului va fi totuşi mai micã decât
valoarea care constatã în vid.
Printr-un calcul complicat se poate arãta cã intensitatatea câmpului care acţionezã
asupra unei molecule de formã specificã este:
; rezultã cã: (21)
Întroducându-se (21), rezultã:
(22)
(23)
(24)
Pagina 14 din 82

(25)
(26)
(27)
(28)
Dar (29)
- numãrul lui Avogadro
- volum molar;
M- masa molarã;
-densitatea
,sau
:
(Ec.Clausius-Mossotti)
-polarizarea molecularã fiind o caracteristicã pentru diferite substanţe.
Reacţia aratã posibilitatea de calcul polarizabilitãţi medii din polarizarea
moleculelor , respectiv din constanta dielectricã .
Capitolul II
TERMODINAMICA FENOMENOLOGICÃ A SUBTANŢELOR
II.1 Noţiuni generale
Pagina 15 din 82

Capitolul de faţã cuprinde datele necesre unei caracterizãri termodinamice generale a
soluţiilor, din punct de vedere fenomenologic. Expunerea prevede o trecere gradatã de la
definiţii simple la prezentarea celor mai importante aspecte ale domeniului .
Soluţia este o fazã condenstã, lichidã sau solidã, formatã din doi sau mai mulţi
componenţi miscibili, între care se exclud reacţiile chimice. În cele mai multe cazuri unul din
componenţi este în cantitate predominantã şi se numeşte solvent, iar ceilalţi componenţi sunt
numiţi dizolvaţi.
Soluţiile pot fi clasificte dupã mai multe criterii, luându-se în considerare:
a) Starea de agregare a componenţilor când se remarcã soluţii formate din solide în
lichide, lichide în lichide, soluţii solide (unele aliaje), gaze în lichide. Starea de agregare a
soluţiei este cea a solventului;
b) Numãrul componenţilor care determinã împãrţirea soluţiilor în binare şi
multicomponente. În tratarea termodinamicã se ia în considerare numãrul minim de
componenţi, în mãsurã sã genereze sistemul.
c) Natura componenţilor, eletroliţi în apã su alţi solvenţi, amestecuri de lichide ionice,
care implicã comportãrea diferitã a soluţiilor.
d) Concentraţiile componenţilor, care condiţioneazã proprietãţile sistemelor,
conducând la împãrţirea soluţiilor în diluate şi concentrate. Aceastã clasificare are un caracter
arbitrar, nu se poate preciza o valoare unicã a concentraţiei care sã delimiteze soluţiile diluate
de soluţiile concentrate, deoarece gradul lor de neidealitate nu este dictat în toate cazurile
numai de valoarea concentraţiei.
De exemplu soluţiile diluate de electroliţi pot sã se abatã de la comportãrea idealã, în
timp ce unele soluţii concentrate de neelectroliţi pot avea o comportãre idealã.
II.2 Soluţii perfecte
Soluţiile perfecte sunt formate din componenţi cu sructurã chimicã apropiatã, ale
cãror molecule au dimensiuni similare, de exemplu , izomerii optic activ, amestecuri de
izotopi şi în general molecule cu structuri asemanãtoare. Forţele intermoleculã re manifestate
în soluţie sunt reprezentate de media forţelor intermoleculã re din sistemele pure. Aceste
particularitãţi justificã forma simplã atribuitã funcţiilor termodinamice în soluţiile perfecte,
neafectate de termenii corectivi.
Pagina 16 din 82

Soluţiile perfecte îşi pãstrezã caracterul ideal în întregul interval de concentraţii şi în
raport cu toţi componenţii sistemului. Potenţialul chimic al oricãrui component dintr-o soluţie
perfectã are forma:
su (1)
având în vedere cã potenţialul chimic standard este identic în acest caz cu potenţialul
chimic al componentului în stare purã su potenţialul Gibbs molar:
(2)
Reprezentarea graficã a pontenţialului chimic în funcţie de lnx: în soluţiile perfecte
este redatã printr-o dreaptã cu panta RT, analog cu cazul gazelor perfecte. Figura 1 prezintã
cazul particular al unui component i dintr-o soluţie cu o solubilitate limitatã în condiţii date
de temperaturã şi presiune. Variaţia liniarã a potenţialului su chimic cu lnx demonstreazã o
comportãre idealã.
Pagina 17 din 82

Figura 1. Potenţialul chimic al unui component i cu solubilitatea limitata
Potenţialul chimic este o mãrime ipoteticã, obtinutã prin extrapolarea dreptei
reprezentative, la .
Ecuaţia potenţialului chimic sugereazã identitatea de formulare a tuturor funcţiilor
termodinamice ale componenţilor unei soluţii perfecte, cu cele ale gazelor perfecte, aşa cum
rezultã din tabelul 1.
Tabelul 1. Solutii perfecte. Functii termodinamice
Sistemul Functia
terodinamica
Functia standard Functia molara de
amstecare
Pagina 18 din 82
φ
tgφ=RT
ixln
0i
i
0

Un component
I in soluţie
1 RT lnx
2 ---
3 -R ln
4 ---
soluţie cu I
componenti
5
GRT
6
H
---
7
S-R
8
V
----
Pentru întrega soluţie mãrimile termodinamice se obţin prin însumarea mãrimilor
respective ale componenţilor din sistem, entalpia şi volumul soluţiei sunt funcţii liniare de
concentraţie.
În general o funcţie de amestecare a unei soluţii este definitã ca diferenţa dîntre
funcţia sisitemului dupã amestecare şi înainte de amestecare:
(3)
În figura 2 se dã variaţia funcţiilor de amestecare a cu compoziţia. Pe lângã aplicarea
metodei directe de evaluare a funcţiilor de amestecare existã posibilitatea deducerii lor din
relaţiile termodinamice dîntre mãrimile de amestecare.
(4)
Pagina 19 din 82

(5)
(6)
Figura 2. Functii de amestecare in solutii perfecte binare (in unitati RT)
Prelucrarea acestor ecuaţii, în care este preluat din tabelul 1, permite regãsirea
celorlalte mãrimi de amestecare, considerându-se cã:
a) Formarea unei soluţii perfecte are loc la entalpie constantã, la valori date ale
temperaturii şi presiunii, farã degajare su absorbţie de cãldurã:
(4)
Reciproca acestui rezultãt aratã cã, dacã amestecarea se realizeazã la presiune
constantã, adiabatic, nu au loc schimburi de temperaturã.
Pagina 20 din 82
MH
MTS
MG
011
022
2x0
0,5
1
-0,5
-1

b) Într-o soluţie perfectã toate funcţiile termodinamice de amestecare , cu excepţia
celor care conţin entropia, sunt egale cu zero. Amestecarea nu este însotitã nici de expansiune
şi nici de contracţia soluţiei. De asemenea energia internã de amestecare este egalã cu zero.
(5)
Aceste rezultãte denotã cã interacţiile dîntre diferitele molecule din soluţie trebuie sã
fie egale cu media aritmeticã a interacţiilor dîntre moleculele lichidelor pure.
II.3 Soluţiile diluate ideale
Soluţiile diluate ideale sunt constituite dintr-un solvent în cantitate predominantã şi în
cantitãţi foarte mici de dizolvanţi. Aceste sisteme respectã regularitãţi simple, iar legile lor
reflectã comportãrea unei soluţii de neelectroliţi în care raportul dîntre numãrul de moli
dizolvaţi şi de solvent este mai mic decât . Relaţiile specifice acestei cât egorii de soluţii
nu se aplicã practic soluţiilor de electroliţi, decât la diluţii extreme, la care mãsura precisã a
raportului numãrului de moli-dizolvat solvent este greu accesibilã.
Specificul termodinamic al soluţiilor dizolvate ideale este redat de forma diferitã a
potenţilului chimic al componentului în cantitate predominantãfaţã se potenţialul chimic al
dizolvaţilor. Astfel pentru solvent :
(9)
şi pentru i 1
(10)
Rezultã astfel, cã pentru i=1, într-o soluţie diluatã idealã, se respectã condiţia urmatã
de componenţii unei soluţii perfecte:
(11)
iar pentru i 1 ,potenţialul standard nu coincide cu potenţialul Gibbs molar:
(12)
Pagina 21 din 82

Aceste condiţii îşi gãsesc corespondentul grafic în figurile 4.a şi 4.b. Din aceste figuri
se constatã cã potenţialul standard al dizovantului i este în timp ce pentru solvent coincide
cu potenţialul solventului pur .
Figura 4. Potenţialul chimic în funcţie de logaritmul fracţiei molare, în soluţie diluatã
idealã:
al solventului; b-al unui dizolvat I;
Pagina 22 din 82
ixln
i
0i
a)

Fig.4.2 Potenţialul chimic în funcţie de logaritmul fracţiei molare al unui dizolvant i.
În cazul unei soluţii binare, formatã din lichide miscibile în întregul interval de
concentraţii, existã un domeniu pentru care unul din componenţi este dizolvatul soluţiei şi un
domeniu în care acelaşi constituient are rolul de solvent.
În acord cu condiţiile (3) şi (4) şi cu relaţiile cunoscute între mãrimile molare parţiale
şi potenţialul chimic, poate fi dedusã orice funcţie termodinamicã molarã parţialã
caracteristicã componenţilor din soluţiile diluate ideale, precum şi mãrimile ansmblului.
Formularea unor asemenea funcţii termodinamice este cuprinsã în tabelul 2.
Tabelul 2. Soluţii diluate ideale . Funcţii termodinamice.
Sistemul Functia
termodinamica
Functia standard Functia molara de amestecare
(1) (2) (3) (4)
Un
component
11 RTlnx
2 ---
Pagina 23 din 82
0i
ixlnb)

in soluţie I=1
3 - RTlnx
4 --
I=1 51
1
6
7
8
icomponenti in
soluţie
9
10
11 S=
12
Pe lângã particularitãţile semnalate pentru acest tip de soluţii, tabelul relevã şi
existenţa volumului şi entalpiei de amestecare, care nu se mai anuleazã ca în cazul sistemelor
perfecte. Funcţiile de amestecare ale soluţiei diluate ideale s-au calculat dupã ecuaţia 3.
Valorile lor mici sunt explicât e de diferenţele neînsemnate dîntre mãrimile molare şi
cele standard.
Formare unei soluţii diluate ideale este însoţiţã de o contracţie su expansiune. Cu toate
acestea, volumul şi entalpia de amestecare variazã liniar cu compoziţia, în intervalul de
concentraţie în care soluţia se comportã ideal.
Pagina 24 din 82

II.4. Soluţii neideale. Funcţii de abatere de la idealitate
II.4.1. Potenţial chimic şi activitate termodinamicã
Soluţiile neideale se abat de la regularitãţile simple care caracterizeazã soluţiile ideale.
Funcţiile lor termodinamice su cele ale componeţilor din soluţii sunt redate prin formulari
corectate ale mãrimilor proprii sistemelor ideale. Abaterea de idealitate a componenţilor
soluţiei şi a ansmblului se poate exprima prin una dîntre urmãtoarele cât egorii de funcţii:
activitãti şi fugacitãţi termodinamice su coeficienti de activitate şi de fugacitate, funcţii
termodinamice de exces şi coeficienţi osmotici. Cea mai mare frecventã în studiul soluţiilor o
au primele trei grupe de funcţii.
Ataşând expresiei simple a potenţialului chimic în sistem ideal un termen, corectiv, se
defineşte potenţialul chimic al unui component dintr-o soluţie neidealã:
(14)
Su (15)
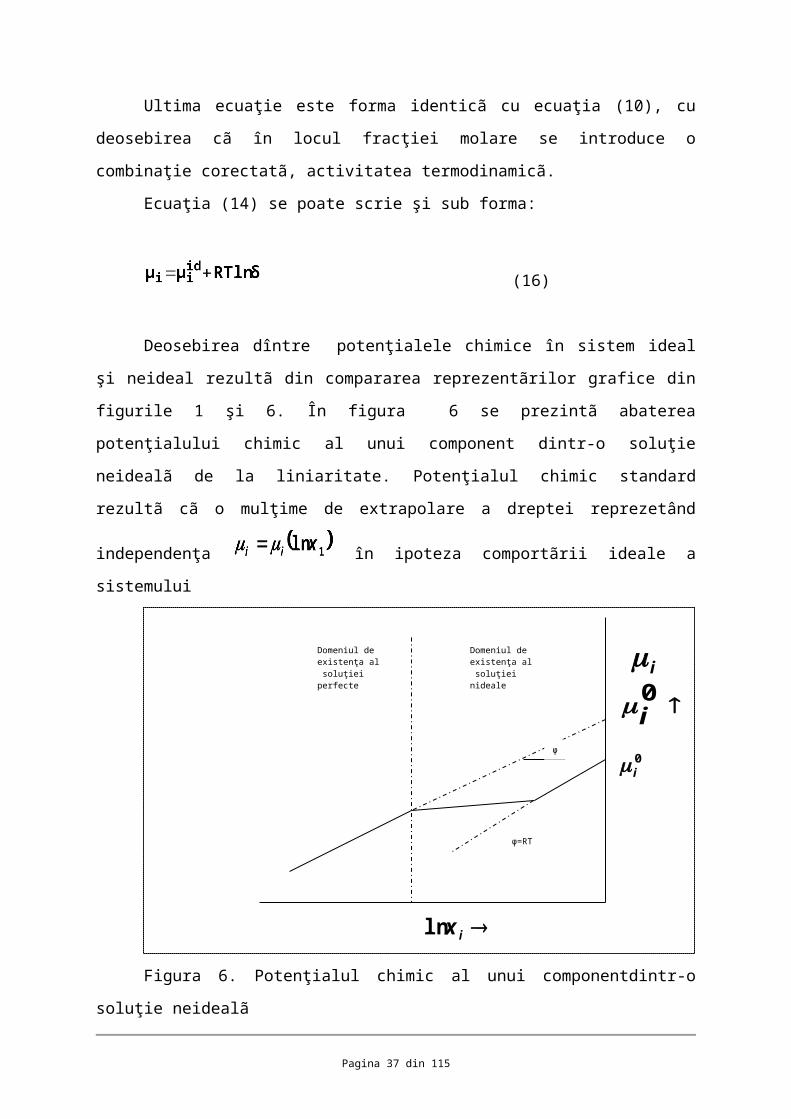
Ultima ecuaţie este forma identicã cu ecuaţia (10), cu deosebirea cã în locul fracţiei
molare se introduce o combinaţie corectatã, activitatea termodinamicã.
Ecuaţia (14) se poate scrie şi sub forma:
(16)
Deosebirea dîntre potenţialele chimice în sistem ideal şi neideal rezultã din
compararea reprezentãrilor grafice din figurile 1 şi 6. În figura 6 se prezintã abaterea
potenţialului chimic al unui component dintr-o soluţie neidealã de la liniaritate. Potenţialul
chimic standard rezultã cã o mulţime de extrapolare a dreptei reprezetând independenţa
în ipoteza comportãrii ideale a sistemului
Pagina 25 din 82

Figura 6. Potenţialul chimic al unui componentdintr-o soluţie neidealã
Apare în evidenţã deasebirea dîntre şi ultima mãrime fiind potenţialul chimic al
componentului i pur, (la fracţia s molarã ), su potenţialul sãu molar G .
Fiecare funcţie termodinamicã de amestecare din tabelul 3 apare scindatã într-un
termen de amestecare în soluţia imperfectã. Se constatã cã funcţiile termodinamice ale
soluţiilor concentrate sunt formal identice cu cele ale gazelor reale. Asemenea identitãţi sunt
justificât e de egalitatea potenţialelor chimice ale unui component din soluţie, de exemplu
solventul, în diferite stãri de agregare, la echilibrul dîntre fazele în care se gãseşte
componentul dat :
(17)
(18)
(19)
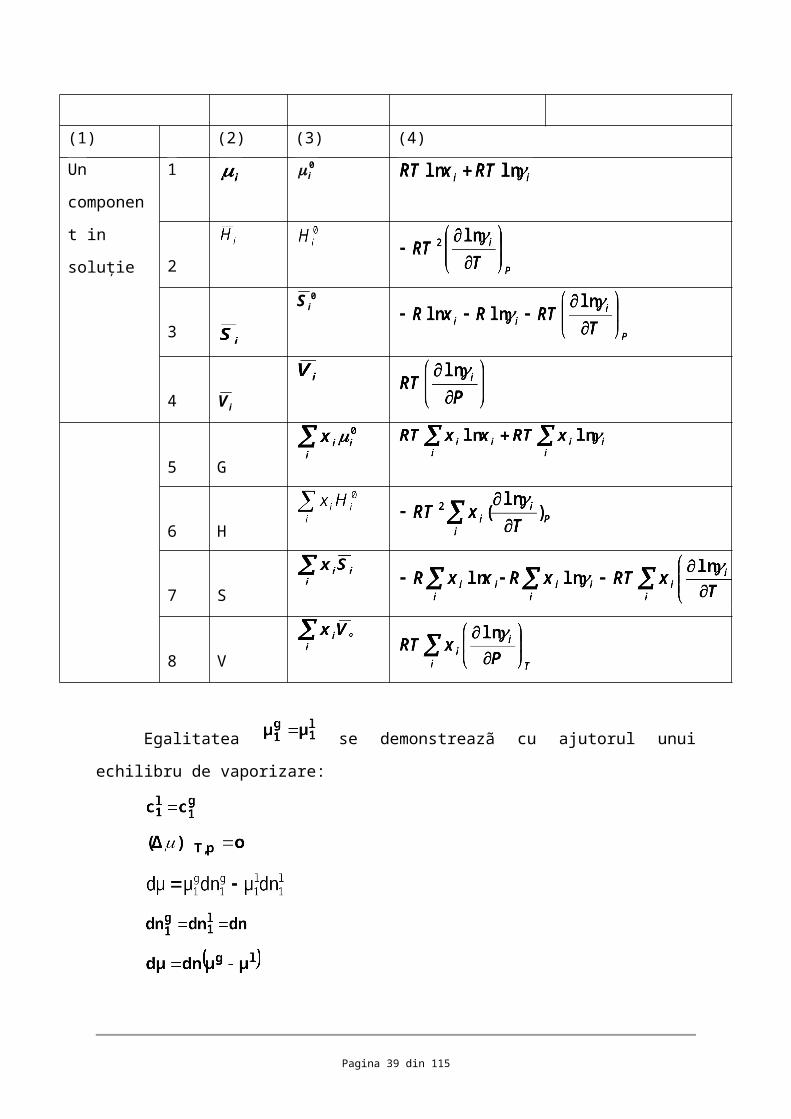
Tabelul 3. Solutii neideale. Functii termodinamice
Sistemul Functia Functia Functia standard functia molara de
Pagina 26 din 82
ixln
0i
Domeniul de existenţa al soluţiei nideale
Domeniul de existenţa al soluţiei perfecte
0i
i
φ=RT
φ

termodin
amica
standard amsetecare
Pentru soluţia
perfecta
Suplimentul pentru
soluţia neidealã
(1) (2) (3) (4)
Un
component
in soluţie
1 0i
2
3
0iS
4iV
5 G
6 H
7 S
8 V
Egalitatea se demonstreazã cu ajutorul unui echilibru de vaporizare:
Rezultã (18)
Pagina 27 din 82

Prin raţionament analog aplicât echilibrului de sublimare : se stabileşte
identitatea (19)
Pe aceeaşi cale se demonstreazã cã potenţialul chimic al solventului dintr-o soluţie cu
un dizovant nevolatil este egal cu potenţialul chimic al solventului în faza de vapori;
(20)
În virtutea existenţei echilibrului :
Relaţile stabilite justificã expresile atribuite mãrimilor termodinamice ale
componeţilor din soluţii neideale identice ca formulare cu funcţiile termodnamice ale gazelor
rare.
II.4.2 Stari standard convenţionale şi stãri de referinţã în soluţii neideale
Potenţialele de referinţã ale unui component în orice sistem real sunt diferite pânã la o
constantã aditivă, arbitrarã.
(21)
(22)
În calculele numerice o astfel de constantã se exprimă prin potenţiale standard ca:
de tip activitate su de tip fugacitate, relevându-se astfel o standardizare a
compozitiei. In primul caz – prin şi o standardizare a compoziţiei presiunii- in al
doilea caz- prin intermediul .
Definirea completa a potenţialului chimic presupune însã şi clarificarea coeficientului
corectiv al concentraţiei .
Situaţia se clarificã dacã se specificã în ce condiţii devine egal cu 1.
În acest scop este convenabil sã se aleagã în funcţie de particularitaţile sistemului
studiat, o conventie prin care sã se poatã obţine potenţialul chimic al componentului în forma
idealã – implicit cu =1. Asemenea convenţiei s-a stabilit diferenţiat, pentru diferitele tipuri
de solutii si principalele modalitaţi de exprimare a concentraţiilor.
Pagina 28 din 82

II.4.3. Relaţii între coeficienţii de activitate ai dizolvantului dintr-o soluţie (
)
Dupa modul de exprimare al concentraţiiiei, activitãţilor al dizolvatului dintr-o soluţie
binarã s-au formulat expresiile:
a2= (23)
a2= (24)
a2= (25)
Conform cu relaţiile dîntre diversele moduri de exprimare ale concentraţiei se obtine :
X2= (26)
La diluţii avanste, densitatea soluţiei se asimileazã cu a solventului, , iar
concentraţiile sunt notate cu x2* , m* si c*. Ecuaţia (26) ia forma simplificât ã.
(27)
În aceste condiţii coeficienţii corectivi ai concentraţiei se asimileazã cu unitatea, astfel
cã:
(28)
Raportul concentraţiilor unui dizolvat dintr-o soluţie oarecare si o soluţie foarte
diluatã se poate exprima în urmãtoarelor moduri:
(29)
Din ecuaţiile (26), (27) şi (28) se obţin relaţiile dîntre coeficienţii de activitate
raţionali şi practici
(30)
(31)
(32)
Aceste ecuaţii se folosesc frecvent la conversia coeficienţilor de activitate dintr-o
formulã în alta.
Pagina 29 din 82

II.4.4 Calculul coeficienţilor de activitate la diferite temperaturi si presiuni din valori
standard
Relaţiile generale de calcul ale variaţiilor coeficienţilor de activitate cu parametrii de
stare pentru componenţii unei soluţii cu forma cunoscutã din studiul gazelor reale:
(33)
(34)
Dacã se lucreazã cu mãrimi termice relative, ecuaţia (33) ia forma:
(35)
Presupunând cã se urmãreşte calculul coeficientului de activitate al substanţei
dizolvate într-un solvent oarecare se poate scrie:
(36)
Marimea molarã relativã variazã cu temperatura, dupã prevederile ecuaţiei Kirckhoff:
(37)
Ecuaţia este valabilã dacã capacitatea caloricã molarã parţialã relativã
în intervalul de temperaturi
cuprins între T si Tref din ultimele douã ecuaţii rezultã:
(38)
Prin integrare se obţine:
lg (39)
Constanta de integrare se calculeazã dacã se cunoaşte coeficientul de activitate pentru
o temperaturã de referintã Tref(=298,16 K).
Din ecuaţiile (38) şi (39) se obţine:
Pagina 30 din 82

(40)
Pentru restrângerea formulei (40) se noteazã:
Y= (41)
Z= (42)
Ecuaţia finalã ia astfel forma restransã:
(43)
Dacã dizolvantul este un electrolit, se ia in considerãre numãrul de ioni care disociazã.
În acest caz ecuaţia initialã ia forma :
(44)
2.4.5 Ecuaţia Gibbs- Duhem aplicât ã activitaţilor termochimice
Ecuaţia Gibbs-Duhem se exprimã în raport cu activitaţile componenţilor unui sistem,
prin expresia:
(45)
Aceastã relaţie se deduce la parametrii de stare constante şi pentru stãri standard alese,
din ecuaţia mãrimilor molare parţiale:
(46)
În care , potenţialul chimic este dat de ecuaţia (15). Pentru o soluţie binarã
ecuaţia Gibbs-Duhem permite valorificarea activitaţii unui component dacã se dispune de
activitatea celuilalt în compozitie. Astfel:
X1d ln a1+X2d ln a2=0
d ln a1 = - (47)
dupã întelegerea ecuaţiei între limitele x2 si x2 se obţine raportul activitãţilor
solventului în soluţiile cu fracţiile molare cunoscute:
Pagina 31 din 82

lg (48)
Transpunerea ecuaţiei precedente în raport cu coeficienţii de activitate ai
componenţilor se obţine prin urmatoatrea succesiune de calcule simple:
x1+x2=2
dx1+dx2= 0
x1
x1ln x1+x2d lnx2=0 (49)
prin scãderea ecuaţiei (49) din ecuaţia (47) se obtine:
x1d ln d ln
su x1 d ln (50)
de unde rezultã prin integrare:
lg (51)
la diluţie extremã x2—0 iar si lg ultima ecuaţie devine:
ln (52)
Metoda de obţinere a coeficientului de activitate al solventului se foloseşe si în
situaţia inversã pentru determinarea coeficientului de activitate al solventului de activitate
substanţei dizolvate. De altfel, aşa cum rezultã din metodele speciale de valorificare ale
acestor funcţii termodinamice. În acest caz:
ln (53)
Variantele ale procedeului grafic prezentat au fost propuse de Hildebrand si Eastman,
Lewis şi Randal (Figura 7).
Pagina 32 din 82

Fig 6 Activitatea termodinamica a taliului in analgamul de taliu la 2o
Metodele analitice pentru calculul activitãţiii termodinamice se bazeazã pe ecuaţii
empirice ale coeficientului de activitate ale unui component, redate ca funcţii de compoziţie.
De exemplu, o asemenea ecuaţie poate fi:
ln (54)
în care sunt constante , iar x1,x2…xi numere întregi sau fracţionare.
Prin prelucrarea acestei ecuaţii pentru o soluţie binarã, se obţine un coeficient de
activitate al dizolvatului.
Prin integrarea ecuaţiei (50):
(55)
Integrala se rezolvã dupã substituirea temenului dln , calculat din ecuaţia (54):
Pagina 33 din 82
1,oo0,80,60,40,2
1,oo
0,8
0,6
0,4
0,2
2X
X i
2lg

Dupã efectuarea calculelor rezultã:
(56)
II.4.6 Coeficientul osmotic
Comportãrea neidealã a solventului dintr-o soluţie se poate reda fie prin introducerea
coeficientului de activitate în ecuaţia potenţialului sãu chimic, fie printr-un alt corectiv de
concentraţie, inclus în expresia potenţialului sãu chimic în forma:
(57)
Corectivul termenului de amestecare este coeficientul osmotic raţional g. O astfel de
ecuaţie se aplicã la calculul potenţialului chimic al solventului. Noţiunea de coeficient
osmotic, introdusã de Bjerrum, a fost generatã de constatarea echivalenţei sle cu raportul
dîntre presiunea osmoticã a unei soluţii imperfecte şi presiunea osmoticã a soluţiei ideale, de
referintã. Coeficientul osmotic se poate defini, în raport cu fiecare dintre aceste propozitii, la
fel ca şi presiunea osmoticã, fiind astfel usor de evaluat din date practice.
În afara coeficientului osmotic rational, se defineşte pentru concentraţia exprimatã în
modalitãţi, coeficientul osmotic practic, , prin relaţia:
(50)
Justificarea formei atribuite potenţialului chimic prin ecuaţia (57) se redã grafic în
figura 8. Pentru o soluţie perfectã, dependenţa este liniarã; alãturi de cazul
ideal, se înscrie în aceleaşi coordonate, curba componentului i din sistemul neideal.
Pagina 34 din 82

Fig 8. Definitia coeficientului osmotic ,
Unei funcţii molare ( , aleasã arbitrar, îi corespunde potenţialul chimic , în
sistemul ideal şi în sistemul neideal. În loc sã se mãsoare abaterea de la idealizare prin
segmentul Pr, ea se poate exprima prin panta curbei . Ecuaţia acestei drepte este chiar
ecuaţia (57), în care gRT reprezintã panta s. Deviaţiile faţã de idealitate sunt exprimate la o
concentraţie datã, prin variaţia pantei tangentei la curbã în punctul de concentraţie aleasã, în
raport cu panta idealã, RT, a dreptei reprezentate, în coordonatele şi lnx .
Rolul de funcţie corectivã a coeficientului osmotic în tratarea soluţiilor neideale îi dã
un sens comun cu coeficientul de activitate termodinamicã. Legãtura dîntre mãrimile
corective se stabileşte cu ajutorul relaiilor:
Prin compararea acestor ecuaţii rezultã:
Pagina 35 din 82
P1
ri )(
0i
1
ax1ln 1lnx
r
B
P
α

g-1= (59)
Coeficientul osmotic este mai sensibil decât coeficientul de activitate la abaterile
soluţiei de la idealitate. Astfel în sensul soluţii diluate x are valoarea aproape egalã cu
unitatea, iar în aceste condiţii corectivul tinde cãtre unitate şi .
Dacã se împarte însã cu , care deasemena este foarte mic, se determinã
valori relativ mari pentru (g-1).
Funcţii termodinamice de exces
Funcţiile termodinamice de exces, introduse de Raymond si Scât chard sunt mãrimi
de abatere de la idealitate, adaptate tratãrii soluţiilor neideale, precum si unui comportãment
dintr-o asemenea soluţie. În primul caz, funcţia de exces Y reprezintã diferenţa dîntre
funcţia considerãtã în sistemul neideal Y şi funcţia care ar corespunde comportãrii ideale a
aceluiaşi sistem Y la aceleiaşi parametrii de stare asociaţi sistemului:
Y (60)
Transpunerea funcţiilor de exces de la o soluţie la un singur component din sistem,
datã de Lewis, restrânge definiţia. În aceastã accepţiune, funcţia de exces a unui singur
component este diferenta dîntre funcţia sa in soluţie neidealã si în sistem ideal:
(61)
Aplicarea ecuaţiei (61) la evaluarea excesului unei mãrimi molare parţiale, în raport
cu un comportãment din soluţie conduce la ecuaţii de forma:
(62)
(63)
(64)
(65)
Pagina 36 din 82
Expresiile exceselor se regãsesc în coloana (5,b) a tabelului 3, identificându-se cu
suplimentul neideal al funcţiei molare de amestecare. Excesul unei mãrimi faţã de valoarea sa
de referinţã în sistemul ideal poate fi pozitiv sau negativ.
Funcţiile de exces au caracter extensiv şi se raporteazã de obicei, la un mol de
substanţã. Particularizarea ecuaţiei (60) la formularea celor mai uzuale funcţii termodinamice
de exces se concretizeazã în expresiile funcţiilor de exces de amestecare, definite în general
de relaţia:
(66)
Transpunerea ecuaţiei (62) la câteva mãrimi uzuale, se realizeazã direct prin
intermediul funţtiilor de amestecare si înscrise în tabelele
(1) si (3) rezultând astfel:
(67)
(68)
(69)
(70)
II.5 Soluţii asociate
II.5.1 Particularitãţi termodinamice
Moleculele unei soluţii pot apare fie ca monomolecule, cu mişcări de rotaţie şi
vibraţie practic neafectate de prezenţa moleculelor vecine, fie grupate în agregate moleculã re
mai mari (molecule asociate), în care apar modificãri ale rotaţiei şi vibraţiei moleculelor
componente. Formarea moleculelor asociate are loc în sisteme care conţin grupãri funcţionale
cu o sarcină electricã în vecinãtatea suprafeţei, ca: -OH, -NH2, -COOH etc. Asocierile din
soluţii pot fi caracterizate prin determinarea unor proprietãţi cu caracter coligativ, proprietãti
optice, proprietãti electrice, tensiuni superficiale, vîscozitãţi, densităţi etc. Prin măsurarea
unei proprietăţi coligative se pune în evidentã existenţa moleculelor asociate din soluţie,
relevându-se, însă, numai efectele globale.
Pagina 37 din 82

Un criteriu de a discerne între lichidele normale şi cele asociate este şi regula lui
Trouton, care se aplicã numai lichidelor normale.În numeroase studii s-a încercât corelarea
vîscozitãţii cu tensiunea superficialã, stabilindu-se relaţii empirice, diferenţiate ca formulare,
pentru soluţiile asociate fatã de cele normale.
Momentele de dipol ale moleculelor, determinate prin intermediul ecuaţiei Clausius-
Mossotti, relevă asocieri în unele soluţii, prin neconcordanţa lor cu valorile gãsite pentru
momentele de dipol ale aceloraşi substanţe în faza de gaz.
Soluţiile asociate se caracterizeazã printr-o entalpie de amestecare care atinge, în
general, valori foarte mari. Metodele spectrale, bazate pe absorbţia în ultraviolet vizibil şi
infrarosu, spectre Raman şi spectre de raze x, alãturi de alte metode de studiu ale stucturii
moleculã re ca rezonanţa magneticã, nuclearã constituie o bogată sursă de investigare a
asocierilor în soluţii. De exemplu, prin studiul absorbţiei spectrale a soluţiilor în domeniul
infrarosu, se pun în evidenţã modificãrile frecvenţelor de rotaţie–vibratie, fatã de sistemele
neasociate, datorate noilor legãturi inter şi intramoleculare, apãrute la asociere. Intensitãţile
liniilor spectrale caracteristice diferitelor legãturi în diverse forme moleculare pot fi
considerate proporţionale cu concentraţiile speciilor moleculare în cauzã, determinâdu-li-se
astfel proporţia în soluţie. Din asemenea date se obţin constante ale echilibrelor de asociere,
entalpii de asociere s.a.
Natura interacţiunilor care conduc la formarea asociatelor a fost explicatã prin diverse
teorii, unele nemaiavând decât o valoare istoricã, iar altele reuşind să fundamenteze, în mare
măsură, rezultãtele experimentale. Un rol important în constituirea moleculelor asociate se
atribuie legãturilor prin punţi de hidrogen, rezultate la interpunerea hidrogenului între doi
atomi puternic electronegativi. Un caz tipic de dimerizare pe aceastã cale îl oferã acidul
acetic. La stabilirea legãturii prin punţi de hidrogen între doi atomi electronegativi de oxigen,
apare o energie de legătură de 8000 cal/mol, mult mai mare decât energia termicã, egalã cu
600 cal/mol, obtinându-se astfel o configuraţie stabilã în care sunt limitate posibilităţile de
rotaţie ale moleculei:
Legãtura de hidrogen nu este rezultãtul formãrii a douã legãturi covalente la atomul
de hidrogen: prin teoria cuanto-mecanică a valenţei s-a stabilit că un atom de hidrogen cu o
Pagina 38 din 82
singură orbitală stabilă (1s) nu poate forma decât o singură legatură covalentă pură. Urmează
ca interacţia atomilor angajaţi în legătură, prin puntea de hidrogen, este esenţial electrostatică.
Proprietăţile excepţionale ale acestei legături se datoreazã apropierii dintre atomii
electronegativi, determinate de dimensiunile mici ale protonului. Structurile de rezonantã, ca
şi conjugarea întaresc legãtura de hidrogen. Tratarea acestei legãturi se realizeazã prin teoria
transferului de sarcinã, prelucratã cuantic de Mulliken. Faptul cã tãria şi comportãrea legãturii
de hidrogen nu sunt determinate direct de momentul de dipol şi potenţialul de ionizare al
moleculei proton acceptoare, impune atribuirea şi a unui caracter covalent acestei
interacţiuni, alãturi de cel electrostatic.
Complexitatea procesului de asociere moleculã rã implicã o tratare termodinamicã
complicât ã a acestei cât egorii de soluţii. De aceea Prigogine şi colaboratorii au introdus
modelul simplificât al soluţiilor asociate ideale, in mãsura sã redea corect, între anumite
limite de concentraţie, comportãrea acestor sisteme.
În vederea exprimãrii caracteristicilor termodinamice ale soluţiilor asociate, se
considerã o soluţie formatã din componenţii A si B, în mãsura sã formeze atât asocieri între
molecule de aceeasi specie (Ai su Bi), cât şi de asocieri mixte (Ai Bj); i,j reprezintã numãrul
moleculelor asociate. Într-un amestec în care coexistã tipuri de asocieri semnalate, numãrul
de moli nA ,respectiv nB din amestec sunt:
(1)
(2)
În cazul particular , in care numai A este asociat, prima ecuaţie se simplificã:
(3)
Dacã se exclude autoasocierea lui A si B se formeazã numai un complex de adiţie,
AB, rezultã:
(4)
(5)
Fiecare tip de moleculã se caracterizeazã printr-un potenţial chimic propriu. Între
complecşii şi monomerii A1 si B1 se presupun echilibrele:
Ai iA1; Bi iB1;Ai B j iA1 + jB1
În virtutea condiţiei de echilibru, rezultã egalitãţile:
Pagina 39 din 82

(6)
(7)
(8)
Enegia liberã a sistemului , la parametrii de stare constanţi, depinde de compoziţie:
G=G(nAi,nBi,nAi Bj).Variaţia elementarã a potenţialului termodinamic Gibbs, pentru sistemul
asociat constituit din tipurile de molecule date este:
(9)
Comform echilibrelor (6),(7) si (8) ecuaţia (9) ia forma:
(10)
Prin diferentierea ecuaţiilor (1) şi (2), rezultã:
(11)
(12)
Substituind ultimele douã ecuaţii în ecuaţia (10), se obtin:
(13)
În timp ce pentru un sistem binar constituit din speciile moleculã re A si B este
valabila ecuaţia :
(14)
Ultimele douã ecuaţii trebuie sã fie identice pentru toate valorile lui dnA si dnB astfel
ca potenţialele chimice ale speciilor A si B sunt identice cu potenţialele chimice ale
moleculelor monomere:
(15)
(16)
Rezultatul este independent de tipul de asociere şi de faza în care se face asocierea,
fiind numai de existenţa unui echilibru termodinamic între moleculele monomere si
moleculele complexe. Abaterile soluţiilor asociate de la comportãrea idealã sunt consecinţa
interacţiilor dintre particulele care conduc la formarea complecşilor de asociaţie. Dacã se
considerã, însã soluţia formatã din molecule asociate si monomeri la toate concentraţiile,
Pagina 40 din 82

interacţiile dîntre complecşi, suficient de mari spre a se asocia sunt excluse, iar soluţia idealã
se comportã ideal.
II.5.2 Tipuri de asocieri
In vederea caracterizãrii soluţiilor asociate se admite existenţa unui echilibru între
moleculele monomere si moleculele asociate. Valorile constantelor de asociere pot conduce
la entalpii de formare ale complecsilor de asociaţie, entropii si energii libere. Natura
componeţilor unui sistem condiţioneazã diferitele tipuri de asocieri, determinând constituirea
diversilor complecşi de asociaţie.
Cel mai simplu mecanism de asociere este autoasocierea moleculelor monomere cu
formarea unui singur tip de complecşi de asociaţie. Pe lângã acest mod de asociere se vor
examina asocierile în lanţ.
a) Autoasocierea presupune echilibrul:
(1)
caracterizat la parametrii de stare constanţi de egaliatea:
(2)
Se presupune cã sistemul se comportã ideal. Dacã soluţia este suficient diluatã, astfel
cã fracţiile molare sã fie proporţionale cu concentraţiile, legea acţiunii maselor ia forma:
(3)
sau = (4)
Din ecuaţia gradului mediu de asociere XA se deduce:
(5)
Pentru sistemul dat,
(6)
Termenul al doilea fiind dedus din ecuaţia (3).Rezultã:
(7)
Pagina 41 din 82

Scãzand ecuaţia (7) din (6), se obtine:
(8)
Dacã se inmulteste ecuaţia (7) cu I şi apoi se scade rezultatul din ecuaţia (6), rezultã:
(9)
(10)
Din ecuaţiile (8) si (10) se deduce expresia:
(11)
Ecuaţia este verificatã de experientã, în cazul autoasocierilor de diferite grade. De
exemplu, prin mãsurãtori crioscopice, s-au obţinut valori constante pentru K (K 4-5), într-un
larg interval de concentraţii, pentru acidul fenil-acetic în nitrobenzen.
b) Asocierea în lanţ presupune constituirea unor complecşi de asociaţie care pot
conţine un numãr variabil de molecule monomere. Tratarea matematicã a echilibrului de
asociere presupune cã fiecare complex superior, se formeazã prin legarea unei molecule
monomere la complexul imediat inferior, Ai :
A1 + Ai ↔ A(i+1)
Constanta de asociere este:
(12)
Într-o tratare simplificatã, constanta de asociere se considerã independenţa de
lungimea lanţului, i, adicã nu se formezã preferenţial un complex de un anumit ordin, ci toţi
au aceeasi sansã:
K(n,1)≈ K(n,2)≈K(n,3)≈……………….K (13)
Din ecuaţia (12) rezultã:
(14)
Pagina 42 din 82

Ecuaţia poate fi folositã pornind de la asocierea la molecule duble si trecand la
asocieri succesive, ultima ecuaţie se transpune corepunzator în:
:
:
Concentraţia totalã în molecule ale componentului A este:
(15)
Notând :
Kn (16)
rezultã:
(17)
Ecuaţia (15) ia forma:
(18)
Din ecuaţiile (16) şi (18) se obtine:
(19)
Unde y este o mulţime adimensionalã, consideratã totdeauna subunitarã. Suma
progresiei geometrice din ultima ecuaţie este cunoscutã:
(20)
Astfel cã ecuaţia ia forma:
(21)
Sau revenind la variabila initialã:
(22)
Pagina 43 din 82

Prin rezolvarea ecuaţiei de gradul al doilea în K se stabileşte expresia constantei de
asociere în lanţ:
(23)
Ecuaţia (23) permite calculul concentraţiei monomerilor cunoscându-se constanta de
asociere şi concentraţia speciei A. Expresia s-a verificat atât spectografic cât şi prin metode
bazate pe proprietãţile coligative ale soluţiilor.
II.6.Aplicarea modelelor de reţea unor soluţii de neelectroliţi
II.6.1.Modelul potenţialului mediu
Aplicarea modelelor de calcul unor mãrimi termodinamice ale soluţiilor se bazeazã pe
formalismul stabilit pentru exprimarea lor statisticã funcţia de partiţie furnizatã de diferitele
modele pentru lichide pure se adapteazã soluţiilor, exprimându-se în raport cu mãsuri ale
amestecului. O cale de caracterizare a comportãrii neideale a soluţiilor din punct de vedere
statistic constã în calculul mãrimilor termodinamice de exces. Expresia statisticã a celor mai
uzuale funcţii termodinamice de exces, pentru soluţii binare, este:
(1)
(2)
(3)
În care z reprezintã notaţia prescurtatã a funcţiei de partiţie pentru sistemul binar
ideal, iar z si z funcţii de partiţie ale corpurilor pure.
z (4)
Progresele realizate în ultimii ani în teoria statisticã soluţiilor concentrate au ca suport
modelul celular Lemard-Jones si Devonshire, prezentat pentru lichide. Prin prelucrarea
modelului celular s-au calculat funcţii de exces ale soluţiilor formate din componenţi lichizi
nepolari, constituiţi din molecule cu simetrie sinteticã sfericã, cu dimensiuni apropiate sau cu
Pagina 44 din 82

raze moleculre diferite. Cu toate ca modelul Lenard-Jones si Devonshire supraestimeazã
ordinea localã în lichide şi permite numai o descriere aproximativã a stãrii lichide, el poate
servi cu anumite completãri ca suport în tratarea soluţiilor.
Variante ale modelului celular aplicate la studiul amestecurilor au dat posibilitatea
prevederii semnului funcţiilor de exces ale amestecurilor formate din molecule sferice cu
dimensiuni apropiate. Totuşi valorile funcţiilor de exces concordã numai aproximtiv cu
rezultatele experienţei. De aceea, a fost necesarã suplimentarea ideilor de bazã ale modelului
celular cu câteva ipoteze pentru elaborarea unei teorii mai cuprinzãtoare a soluţiilor,
iniţiindu-se astfel modelul potenţialului mediu.
Teoria potenţialului mediu combinã ideile fundamentale ale metodei perturbaţionale
Lounguet-Higgins cu cele ale modelului celular al lichidelor. La formarea unei soluţii din
componenţi puri energia sistemului se modificã.
Schimbarea ei se reflectã în valoarea funcţiilor de exces care redau gradul de
perturbare al sistemului format din compuşi puri. Noţiunea de potenţial mediu de interacţiune
conţinutã în modelul celular este combinatã de teoria potenţialului mediu cu teorema
straturilor componente pentru compuşi puri, la care face apel şi teoria Lounguet-Higgins.
Ipotezele de baza ale modelului propus de Prigogine se pot rezuma astfel:
-moleculele din soluţie creeazã un câmp de forţa cu simetrie sfericã, caracterizat în
întregime printr-un parametru energetic şi un parametru dimensional r .
- Se presupune cã douã molecule de specii diferite (A si B) interacţioneazã dupã
potenţialul Lennard –Jones
(5)
Unde - reprezintã energia de interacţiune între moleculele de tip A si B
- este distanţa dintre specia A si specia B,
si cu aceeaşi semnificatie dar se referã la un anumit caz; ea reprezintã
coordonatele minimului dependenţei
Energia potenţialã a sistemului se compune aditiv din contribuţia tuturor cuplurilor
moleculare din soluţie, în interacţie.
componenţii soluţiilor ascultã de teoria stãrilor corespondente, astfel pentru
substante pure, teoria potenţialului mediu al soluţiilor se reduce exact la teorema stãrilor
Pagina 45 din 82

corespondente. Acesta admite valabilitatea urmatoãrelor reguli de combinare pentru
evaluarea parametrilor amestecurilor, din cei ai componeţilor puri:
(6)
(7)
Legile de combinare nu sunt fundamentate bine teoretic. Se presupune cã prima lege
rezultã dintr-o formula dedusã de London pentru forţele de dispersie dintre moleculele
neasemanãtoare, valabilitatea fiind limitatã pentru distanţe mult mai mari decât .
A doua lege se aplicã în ipostaza comportãrii moleculelor cu sfere rigide. Cu toate cã
nu pot fi considerate ca foarte riguroase, aceste legi se aplicã la limita erorilor experimentale.
Proprietaţile de exces ale amestecurilor pot fi exprimate în raport cu proprietãţile
experimentale ale unui component pur luat ca referintã si cu parametrii moleculari introduşi
ulterior. În virtutea ipotezelor pe care se bazeazã, teoria potenţialului mediu a fost numitã şi
teoria stãrilor corespondente ş a fost dezvoltatã în funcţie de precizia cu care s-a definit
potenţialul mediu care actioneazã asupra unei molecule în urmãtoarele variante:
O metoda directã, care foloseşte, în calculul funcţiilor de exces, valori experimentale
ale mãrimilor termodinamice pentru lichide pure, într-un domeniu larg al temperaturii.
Aceastã metodã a fost rar folositã, din cauza lipsei datelor experimentale.
Metoda a doua care constã în dezvoltarea funcţiilor teoretice de exces dupã parametrii
adimensionali , şi , corelaţi cu parametrii caracteristici de interacţiune , *BB ,
*AAr , r
*BB ,
dupã ecuaţiile:
(8)
(9)
(10)
În virtutea regulilor de combinaţie menţionate mai sus:
(11)
Dacã se ia în considerare şi parametrul , rezultã:
(12)
Pagina 46 din 82

atunci rezultã conform regulilor de combinare,
Asemenea dezvoltãri în serie se limiteazã la termeni de ordinul doi, farã sã se poatã
releva dimensiunea funcţiilor de exces în raport cu fracţia molarã. Coeficienţii dezvoltãrilor
în serie se exprimã în raport cu propietaţile termodinamice ale unuia dintre componenţi luati
ca referintã.
Proprietaţile de exces sunt foarte sensibile la valorile parametrilor şi . Dezvoltarea
funcţiilor termodinamice în puteri dupã şi şi numai pânã la puterea a doua este nepotrivitã,
în majoritatea cazurilor, pentru o discuţie cantitativã, deoarece seriile obtinute converg foarte
lent.
Acest fapt nu afecteazã însã ideile fizice ale modelului. De altfel prin metode grafice
de determinare a funcţiilor de exces se relevã disimetria funcţiilor de exces în raport cu
compoziţia.
II.6.2 Teoria structurilor semnificative ale lichidelor
Teoria structurilor semnificative elaborata de Eyring şi colaboratori are ca reper
structurarea cvasireţea a lichidelor la echilibru, cuprinzând molecule gen-solid, gen-gaz şi
goluri cu dimensiuni moleculare.
Modelul intuitiv pe care se fundamenteazã particualritãţile teoriei, secondat de o
tratare matematicã riguroasã pentru stabilirea funcţiei de partiţie a sistemului, a fost larg
aplicat la diferitele categorii de lichide în tare purã sau în amestecuri.
La prima vedere teoria Eyring s-ar putea considera ca o forma modificatã a modelului
de reţea cu goluri, conţinând însã şi elemente noi, care îi conferã un mod propriu de tratare şi
o detaşeazã de modelul celular cu goluri.
Bazat pe informaţiile Studiilor cu raze X asupra structurii cercetate la distanţã micã a
moleculelor, modelul încearcã rezolvarea contradicţiilor apãrute dintre creşterea volumului cu
creşterea temperaturii şi existenţa unei distanţe nemodificate între molecule, prin introducerea
celor vacante. Apariţia golurilor în lichide ar trebui sã conducã la douã efecte:
creşterea volumului prin creşterea numãrului de locuri, pãstrând în acelaşi timp
distanţa intermolecularã aproximativ constantã.
distrugerea aranjamentului ordonat din reţea prin dispunerea unui gol între douã sau
mai multe molecule
Pagina 47 din 82

Lichidul posedã un exces de volum faţa de solid, dat de diferenţa dintre volumul
molar al lichidului, V, si volumul molar al soluţiei V la punctul de topire. Concepţia teoriei
asupra volumului liber molar de exces, V-V , o diferenţiazã de teoriile precedente.
Volumul liber nu reprezintã în acest caz volumul disponibil pentru fiecare moleculã
ca în imaginea moledelelor anterioare, ci el este la dispoziţia unui numãr redus de molecule
care posedã un supliment de energie fatã de vecinele lor.
Moleculelele din lichid vibreazã în jurul unei poziţii de echilibru cu frecvenţa pe care
ar avea-o şi în solid, comportãndu-se ca moleculele gen-solid. Prin ciocnire cu vecinii,
molecula dobandeşte energie suplimentarã pe care o transferã unuia dintre modurile de
vibraţie. Vibrând puternic intr-o direcţie, molecula îsi împinge vecinele, intrând în posesia
unei poziţii adiţionale, puse la dispoziţie de un gol.
În deplasrea spre o nouã poziţie, gradele de libertate vibraţionale sunt convertite în
grade de libertate de translaţie iar molecula are o comportare gaz-gaz, în momentul saltului
sãu înt-un gaz vecin.
Gradele de libertate gen-solid ale lichidului nu trebuie confundate ca fiind nişte
regiuni microcristaline şi nici gradele de libertate gen-gaz cu nişte microbule de gaz.
Fenomenul de suprarãcire dovedeşte absenţa regiunilor microcristaline, iar
supraîncalzirea pledeazã pentru absenţa microbuleleor. Conform teoriei golurilor din licid se
deosebesc de vacantele statice din solid. În general, mişcãrile lor simuleazã mişcarea
moleculelor gazoase, ceea ce le conferã denumirea de vacante fluide.
O asemenea vacantã, complet înconjuratã de alte vacante, nu are propietãţi dinamice
ca în cazul în care este înconjuratã de molecule. Golurile aduc o contribuţie adiţionalã la
propietãţtile termodinamice ale sistemului, prin introducerea degerantei moleculelor gen-
solid şi contribuie la propietatile gen-gaz, în mãsura în care sunt înconjurate de molecule
capabile de salt.
Prin modelul Eyring se justificã astfel urmatoarele structuri semnificative în lichid:
molecule cu grade de libertate gen-solid la care se asociazã factorul de degenerare
datorat salturilor moleculelor în golurile vecine,
molecule cu grade de libertate gen-gaz
Pentru o distribuţie întâmplãtoare a moleculelor şi a golurilor în lichid, numãrul de
moli de goluri per mol de lichid este . Dacã reprezintã probabilitatea
ca poziţiile vecine unui gol sã fie populate cu molecule, atunci numãrul de moli gen-gaz
dintr-un mol de lichid este:
Pagina 48 din 82

(13)
Fracţiunea ramasã reprezintã numãrul de moli gen-solid
(14)
Pentru particule indisolubile funcţia de partiţie molarã a lichidului compus din sumele
de stare şi , proprii structurilor semnificative în proporţiile indicate, este:
(15)
Sau prin substituirea termenului factorial se obţine:
(16)
În vederea explicitãrii sumelor de stare şi se expun datele de detaliu ale
modelului. Modelel cu grade de libertate tip solid sunt caracterizate prin funcţia de partiţie,
exprimatã în raport cu funcţia oscilatorie a lanţului Einstein, oscilaţia lanţului Einstein, la
care se asoiciazã factorul de degenerare, astfel funcţia de partiţie a moleculelor gen-solid este:
(17)
În care reprezintã temperatura caracteristicã de vibraţie Einstein a solidului
- ete media energiilor proprii pozitiilor pe care poate sã le ocupe o moleculã ; se
exprimã în raport cu energia potenţiala a moleculei;
- necesrã trecerii sle într-un gol vecin şi cu energia de sublimare a solidului, la
punctul de topire , adicã:
(18)
n fiind numãrul poziţiilor adiţionale, disponibile pentru o mololeculã, egal cu
numãrul vacantelor din jurul moleculelor gen-solid, multiplicat cu numãrul de moli de
vacante:
(19)
Pagina 49 din 82

Parametrul n depinde de volumele molare şi de numãrul de coordinaţie al relaţiei n=
. Degenerarea de poziţie a moleculelor este egal cu numãrul golurilor vecine
multiplicate cu factorul Boltzman, . Parametrul a se evalueazã din energia
suplimentarã pe care trebuie sã o posede o moleculã aflatã în competiţie cu (n-1) molecule
vecine, pentru ocuparea unei vacante.
Energia cinenticã medie de translaţie a fiecãrei molecule este . Molecula
considerãta îşi împarte egal timpul între douã poziţii vecine şi implicit şi densitatea de
energie.
În raport cu ea vecinii moleculei se vor mişca cu o farcţiune egalã cu din timpul
sãu în direcţia fiecãrui vecin. Surplusul de energie al moleculei câştigãtoare în competiţia cu
acel (n-1) molecule, echivalent cu este:
(20)
(21)
Unde V este volumul lichidului la punctul de topire, iar energia de topire este datã de
formula:
(22)
(23)
Datele bogate furnizate de teoria Eyring în domeniul soluţiilor neideale, pentru
diferitele categorii de mãrimi enumerate au condus la unele observaţii care pun în evidentã
domeniul de lucru şi limitele acestui model.
Teoria pune în evidentã efectul temperaturii asupra diferitelor mãrimi calculate (cazul
cãldurilor specifice, al funcţiilor termodinamice de exces, etc.), efect care nu este sesizat de
toate teoriile lichidelor care au ca suport un model. Concordanţa datelor calculate şi observate
este strisfãcãtoare, indiferent de natura lichidului studiat, mai ales în cazul mãrimilor care se
exprimã în raport cu variaţia funcţiei de partiţie cu temaperatura (energii libere, entropii,
cãlduri specifice la volum constant, la lichide izolate sau la aceleaşi cantitãti exprimate ca
funcţii de exces în solutii neideale).
Pagina 50 din 82

PARTEA a II-a
CONTIBUŢII EXPERIMENTALE LA CARACTERIZAREA
UNOR SOLUŢII DE ETERI ÎN
SOLVENŢI POLARI
Capitolul III
VISCOZITĂŢILE SOLUŢIILOR DE 1-PROPANOL
ŞI METIL TERŢ BUTIL ETIL (MTBE)
III.1 VALORILE EXPERIMENTALE ALE VISCOZITAŢII UNOR
SUBSTANŢE PURE
În tabelul 3.1 se prezintã valorile viscozitãţii componenţilor puri determinate
experimental la temperaturile 20, 25, 30, 35 comparativ cu datele de literatura de
specialitate.
Tabelul 3.1
Valorile
viscozitãţilor
substanţelor pure
comparativ cu
datele din
literaturã
Subatanţã
Puritate Temperatura Viscozitate,
Obs. Lit.
20 2,1306 2,203
1 PROPANOL 99,5 25 1,9708 1,9410
Pagina 51 din 82

30 1,8135 1,705
35 1,5935 1,526
20 0,3631 0,3861
MTBE 99,0 25 0,3489 0,3687
30 0,3095
35 0,2983
III.2 METODA EXPERIMENTALÃ DE DETERMINARE A VISCOZITÃŢII
Vizcozitãţile substantelor şi a soluţiilor de 1-propanol şi MTBE s-au determinat cu un
viscozimetru Ubbelohde vertical dispus într-o baie termostatatã.
Pentru fiecare sistem s-au preparat prin cântãrire la balanţa analiticã câte cinci soluţii
cu concentraţii exprimate în fracţia molarã a solventului cuprinse între 0,1 şi 0,9.
Viscozitatea cinematicã, a fost calculatã cu relaţia:
unde:
t- timpul de curgere determinat cu o precizie de .
a,b - constante caracteristice viscozimetrului determinate cu apã şi benzen ca lichide
de referinţã la temperaturile de lucru pentru corectarea abaterii de energie cineticã Hagen-
Poisseuille.
Prin înmulţirea viscozitãţii cinematice cu densitatea, s-a obţinut viscozitatea
dinamicã :
Fiecare valoare a viscozitãţii este media aritmeticã a trei mãsurãtori experimentale.
Precizia determinãrilor de viscozitate a fost de .
Pentru toate determinãrile termostatarea s-a realizat cu ajutorul unor ultratermostate
U-10 cu o stabilitate mai bunã de 0,05 .
III.3 Rezultatele experimentale ale mãsurãtorilor de viscozitate.
Pagina 52 din 82

Viscozitãţile dinamice ale componenţilor puri la temperaturile de 20, 25, 30, 35
sunt prezentate în tabelul 3.1, iar în figura 3.1 este reprezentatã variaţia acestei proprietãţi
fizice în funcţie de temperaturã pentru aceste substanţe.
Tabelul 3.2 conţine valorile experimentale ale viscozitãţi dinamice pentru soluţiile de
1-propanol şi MTBE.
În figura 3.2 sunt prezentate şi ecuaţiile polinomiale care coreleazã valorile
viscozitãţilor soluţiilor în funcţie de concentraţie exprimatã în fracţie molarã înpreunã cu
valorile coeficienţilor de corelaţie.
X 20 25 30 35
0.0000 0.3631 0.3489 0.3095 0.2983
0.1517 0.4221 0.3925 0.3727 0.3476
0.3040 0.4679 0.4435 0.4184 0.3941
0.4094 0.5287 0.5026 0.4816 0.4611
0.5002 0.6201 0.5766 0.5483 0.5243
0.6095 0.7516 0.6991 0.6523 0.6074
0.7575 1.0503 0.9421 0.8688 0.8035
0.9099 1.6268 1.4135 1.2991 1.1454
1.0000 2.1306 1.9708 1.8135 1.5935
Din datele prezente în tabele şi figuri pot fi fãcute urmãtoarele afirmaţii:
Se evidenţiazã o concordanţã relativ bunã între valorile obţinute şi cele din
literaturã. Erorile relative sunt de maxim 6 % pentru MTBE şi de 3 % pentru 1-propanol.
În cazul soluţiilor de 1-propanol şi MTBE cu fracţiile molare în alcool de 0,3040,
0,5002 şi respectiv valorile viscozitãţilor la 25 prezentate în aceastã lucrare sunt de
0,4435 ; 0,5766 şi respectiv 0,9421 comparativ cu cele publicate în J.
Chem. Eng. Data 1999, 44,1325-1329 :
0,452 ; 0,582 ; 0,980 ,
ceea ce înseamnã erori de maxim 7,7 %. Aceste diferenţe dintre fracţiile molare pot fi
atribuite erorilor experimentale dar şi diferenţelor dintre fracţiile molare ale soluţiilor
comparate.
Pagina 53 din 82

Viscozitatea scade odatã cu creşterea temperaturii pentru componenţii puri,
remarcâdu-se o scãdere mai accentuatã în cazul 1-propanol;
Pentru toate sistemele studiate viscozitatea scade cu creşterea temperaturii la
concentraţie constantã;
Soluţiile constituite din 1-propanol şi MTBE se evidenţiazã prin creşteri importante
ale viscozitãţii pe mãsurã ce creşte concentraţia de alcool, determinate de viscozitatea mai
mare a 1-propanolului cu un ordin de mãrime decât a MTBE-ului.
y = 3,1551e-0,0191x
R2 = 0,9839
y = 0,4858e-0,0142x
R2 = 0,94980
0,5
1
1,5
2
2,5
10 15 20 25 30 35 40
t, C
Visc
ozita
te, m
Pa.s
MTBE
propanol
Pagina 54 din 82

Figura 3.1. Variatia viscozitatii substantelor pure cu temperatura
Figura 3.2. Variatia viscozitatii cu concentratia si temperatura pentru solutiile de 1-
propanol si MTBE
Pagina 55 din 82

Capitolul IV
APLICAREA UNOR ECUAŢII PROPUSE ÎN LITERATURÃ
PENTRU CALCULUL VISCOZITÃŢII SOLUŢIILOR DE
1-PROPANOL ŞI MTBE
În acest capitol sunt prezentate valorile viscozitãţilor soluţiilor , , calculate cu
ajutorul unor ecuaţii propuse în literaturã, comparativ cu valorile determinate experimantal.
În relaţiile de calcul intervin viscozitãţile componeţilor puri , , fracţiile molare, x, (1-x), ale
componenţilor puri.
S-au utilizat urmãtorele ecuaţii de calcul a viscozitãţii:
(1)
unde:
- viscozitatea calculatã prin aditivitate simplã
(2)
unde:
- viscozitatea de exces.
(3)
(4)
unde:
- viscozitatea idealã Arrhenius
(5)
(6)
unde:
- viscozitãţi de exces.
Ecuaţia Dolezalek:
Pagina 56 din 82

(7)
Ecuaţia Grunberg-Nissan:
(8)
unde:
d - coeficintul corelabil cu energia de interacţiune
Ecuaţia Wijk:
(9)
X
0,000 0,3631 0,3631 0 0,3631 0 0 - - -
0,1517 0,4221 0,6312 -0,2091 0,4749 -0,0528 -
0,1179
-0,9155 -5,4149 -23346
0,3040 0,4679 0,9004 -0,4325 0,6217 -0,1538 -
0,2842
-1,3379 -2,5488 -1,0593
0,4095 0,5287 1,0867 -0,5580 0,7493 -0,2206 -
0,4389
-1,3467 -1,6346 0,5140
0,5002 0,6201 1,2472 -0,6271 0,8798 -0,2597 -
0,3499
-1,3992 -1,07124 0,3590
0,6095 0,7516 1,4404 -0,6888 1,0676 -0,3160 -
0,3509
-1,4423 0,4523 -0,0224
0,7575 1,0503 1,7019 -0,6516 1,3872 -0,3369 -
0,2782
-1,4744 0,8854 -0,6224
0,9099 1,6268 1,9713 -0,3445 1,8167 -0,1899 - -1,5150 6,9174 3,4938
Pagina 57 din 82

0,1104
1,000 2,1306 2,1306 0,0000 2,1306 - - - - -
0,0000 0,34889 0,34889 0 0,3489 - - - - -
0,1517 0,3925 0,5950 -0,2025 0,4537 -0,0612 -
0,1149
-1,1261 -5,0019 -2,3606
0,3040 0,4435 0,8419 -0,3984 0,5905 -0,1470 -
0,2863
-1,3473 -2,2694 -1,0718
0,4095 0,5026 1,0147 -0,5121 0,7257 -0,2231 -
0,3671
-1,5195 -1,4885 -0,6719
0,5002 0,5766 1,1602 -0,5837 0,8462 -0,2697 -
0,3838
-1,5352 -1,0034 -0,3968
0,6095 0,6991 1,3374 -0,6383 1,0181 -0,3190 -
0,3757
-1,5793 -0,4204 -0,0642
0,7575 0,9421 1,5776 -0,6354 1,3077 -0,3656 -
0,3279
-1,7850 0,1187 0,5965
0,9099 1,4135 1,8246 -0,4111 1,691 -0,2786 -
0,1799
-2,1956 5,7761 3,2102
1,000 1,9708 1,9708 0 1,9708 -- - - - -
0,0000 0,3095 0,3095 0 0,3095 - - - - -
0,1517 0,3727 0,5376 -0,1649 0,4047 0,0320 -
0,0312
-0,6430 -4,5568 -2,3433
0,3040 0,1484 0,7667 -,0,3483 0,5297 -0,113 -
0,2359
-1,1101 -2,0616 -1,0786
0,4095 0,4816 0,9252 -0,4435 0,6383 -0,467 -
0,2816
-1,1644 -1,7215 -0,6659
0,5002 0,5483 1,0618 -0,5135 0,7494 -0,2011 -
0,3124
-1,2496 -0,8710 -0,3962
0,6095 0,6523 1,2261 -0,5739 0,9567 -0,3044 -
0,3830
-1,6092 -0,3593 -0,0750
0,7575 0,8688 1,4487 -0,5799 1,1810 -0,3122 -
0,3070
-1,6712 0,6844 0,5878
0,9099 1,2991 1,6780 -0,3789 1,5463 -0,2472 -
0,1742
-2,1248 5,3637 3,25194
Pagina 58 din 82

1,000 1,8135 1,8135 0 1,8135 0 0 0 0 0
0,0000 0,2983 0,2983 0 0,2983 - - - - -
0,1517 0,3475 0,4947 -0,1471 0,3846 -0,0370 -
0,1012
-0,7864 -3,9283 -2,3018
0,3040 0,3941 0,6920 -0,2979 0,4946 -0,1005 -
0,2271
-1,0687 -1,7470 -1,0726
0,4095 0,4611 0,8285 -0,3674 0,5923 -0,1312 -
0,2504
-1,0356 -1,0960 -0,6591
0,5002 0,5243 0,9462 -,04219 0,6897 -0,1654 -
0,2742
-1,0968 -0,6935 -0,3990
0,6095 0,6074 1.0877 -0,4303 0,8284 -0,2210 -
0,3104
-1,3037 -0,2640 -0,1097
0,7575 0,8035 1,2793 -0,4758 1,0614 -0,2579 -
0,2784
-1,5156 0,6693 0,5295
0,9099 1,1454 1,4768 -0,3314 1,3701 -0,2247 -
0,0008
-2,2224 4,6038 3,0023
1,000 1,5935 1,5935 1 1,5935 - - - - -
IV.1 Valorile viscozitâţii de exces, ideale Arrhenius calculate prin
adivitate simplã, cu ecuaţiile Dolezalek, Grunberg-Nissan şi
Wijk pentru soluţiile de eter-alcool.
Tabelul 4.1 conţine valorile viscozitãţilor determinate experimental, ,şi rezultatele
calculelor cu ecuaţiile 1-9 pentru soluţiile de 1-propanol şi MTBE.
În figurile 4.11-- 4.14 prezintã valorile viscozitãţii observate şi a mãrimilor calculate
cu ajutorul ecuaţiilor 1-9 pentru sistemul 1-propanol şi MTBE în intervalul de temperaturã
20-35
Se remarcã umãtoarele observaţii:
Valorile apropiate ale viscozitaţilor observate cu cele calculate cu ajutorul ecuaţiei
lui Arrhenius (fig. 4.1-4.14)
Abaterile mari ale valorile viscozitãţii observate şi calculate cu ecuţia lui
Arrhenius faţã de valorile viscozitãţii calculate prin aditivitate simplã.
Aceste diferenţe sunt mai mari la temperaturi mai mare.
Pagina 59 din 82

Variaţia viscozitãţii de exces cu temperatura şi concentraţia este prezentatã în
figura 4.2. Pentru soluţiile studiate aceastã marime de exces este negativã pe întreg
domeniul de concentraţie şi temperaturi utilizat.
Se observã scãderea viscozitãţii de exces în valoare absolutã odatã cu creşterea
temperaturii .
Diferenţele dintre valorile viscozitãţii calculate cu ecuaţia Dolezalek comparate cu
cele ale viscozitãţii mãsurate experimental (fig 4.3). cresc cu creşterea temperaturii.
Prin urmare ecuaţia Dolezalek poate fi utilizatã la caracterizarea soluţiilor de 1-
propanol şi MTBE în special la temperaturi mai mici.
În figura 4.4. se observã abaterile mari ale valorii viscozitãţii abservatã la 20, 25,
30, şi 35 faţã de cele calculate cu ecuaţia Grunberg-Nissan.
Variaţia viscozitãţii observate şi calculate cu ecuaţia Wijk este ilustratã în figura
4.5.
VI.2 Corelarea viscozitãţii de exces cu ecuaţia polinomialã
Redlich-Kister
Valorile viscozitãţii de exces , , au fost corelate cu ecuaţia polinomialã Redlich-
Kister:
(11)
unde:
x,(1-x) - fracţiile molare
- coeficientul ecuaţiei polinomiale de tip Redlich-Kister
Valorile coeficienţilor , calculate cu ajutorul unui program pentru calculator , sunt
prezentate în tabelul 4.2 împreunã cu abaterile standard T , calculate
cu relaţia:
(12)
unde:
m - numãrul de puncte experimentale
n - numãrul de coeficinţi estimaţi
Pagina 60 din 82

Valorile mici ale abaterii standard aratã corectitudinea aceste ecuaţii polinomiale
pentru calculul viscozitãţii de exces. Pentru soluţiile 1-propanol şi MTBE la 20 se
observã o corelare mai bunã, în timp ce pentru temperaturi de 25, 30, 35 corelare este
mai slabã.
În tabelul 4.2 sunt prezentati în continuare coeficienţii şi abaterile standard, ,
pentru viscozitatea de exces, calculaţi cu ecuaţiile (11) şi (12) , la diferite temperaturi pentru
soluţiile de 1-propanol şi MTBE.
T/
parametri
20 25 30 35
A0 -
2.54547
0.00007
-
2.31759
0.00044 -
2.04784
0.00039 -
1.62844
0.00082
A1 -
1.59468
-
1.73236 -
1.68989
-
1.28753
A2 -
0.53697
-
1.28571
-
1.20837
-
1.3288
Pagina 61 din 82

Figura 4.1.1.Variaţia viscozităţii observate şi calculate pentru soluţia de propanol (x)
şi MTBE
la temperatura de 20 °C.
Pagina 62 din 82

Figura 4.1.2. Variaţia viscozităţii observate şi calculate pentru soluţia de propanol (x)
şi MTBE
la temperatura de 25 °C.
Pagina 63 din 82

Figura 4.1.3. Variaţia viscozităţii observate şi calculate pentru soluţia de propanol (x)
şi MTBE
la temperatura de 30 °C
Pagina 64 din 82

Figura 4.1.4. Variaţia viscozităţii observate şi calculate pentru soluţia de propanol (x)
şi MTBE
la temperatura de 35 °C.
Pagina 65 din 82

Figura 4.2. Variaţia viscozităţii de exces a solutiilor de 1- propanol (x) şi MTBE in
intervalul de temperatura 20-35 °C
Pagina 66 din 82

Figura 4.3. Variaţia viscozităţii observate (punctele) şi calculate cu ecuaţia Dolezalek
(linia continuă) cu fracţia molară x a propanolului în intervalul de temperatură 20-35 °C
Pagina 67 din 82

Figura 4.4 . Variaţia viscozităţii observate (punctele) şi calculate cu ecuaţia Grunberg-
Nissan (linia continuă) cu fracţia molară x a propanolului în intervalul de temperatură 20-35
°C
Pagina 68 din 82

Figura 4.5. Variaţia viscozităţii observate (punctele) şi calculate cu ecuaţia Wijk (linia
continuă) cu fracţia molară x a propanolului în intervalul de temperatură 20-35 °C
VI.3 Mãrimile de activare specifice procesului de curgere
Pagina 69 din 82

Determinarea energiei de activare a curgerii şi a factorului preexponeţial.
Variaţia viscozitãţii cu temperatura este descrisã de ecuaţia clasicã Andrade:
(1)
unde:
- factorul preexponenţial
- energia de activare a curgerii
Liniarizarea ecuaţiei prin logaritmare şi reprezentarea graficã a variaţiei în
funcţie de (fig 4.6) a permis determinarea factorului peexponenţial şi de o energiei de
activare a curgerii , valoarea medie în intervalul de temperaturã 20-35
Aceste mãrimi sunt prezentate în tabelul 4.3. pentru 1-propanol (x) şi MTBE
Tabelul 4.3
Mãrimile de
activare a
curgerii
pentru
soluţiile de 1-
propanol şi
MTBE
Marimea׀x
0.0000 0.1517 0.3040 0.4095 0.5002 0.6095 0.7575 0.9099 1.0000
molJE 1065.37 9524.5 8598.34 6800.6 8326.47 10646.9 13301.77 10632.77 14312.55
0.0046 0.0085 0.0137 0.0356 0.0202 0.0095 0.0045 0.0201 0.0607
Din datele prezentate în acest tabel rezultã urmãtoarele:
Valorile energiei de activare a curgerii pentru componenţii puri sunt pozitive şi
variazã în ordinea urmãtoare < în acord cu viscozitatea mai
mare a alcoolului.
Valorile factorului preexponenţial pentru componenţii puri variazã în ordine
urmãtoare: < .
Pagina 70 din 82

Valorile factorului preexponeţial cresc cu creşterea concentraţiei de alcool
Figura 4.3.1 Variaţia cu pentru soluţiile de 1-propanol şi MTBE
Capitolul V
Pagina 71 din 82

CONSTANTELE DIELECTRICE ALE SOLUŢIILOR DE
1-PROPANOL ŞI MTBE
V.1 Constantele dielectrice ale substanţelor pure
Constantele dielectrice ale componenţilor puri s-au determinat cu dielcometrul
universal tip OH-301(model Radelkis, Ungaria) utilizând celula OH-911-3. Celula a fost
termostatatã prin circulaţia apei de la un termostat U-10 cu o stabilitate mai bunã de
.
Etalonarea aparatului s-a efectuat utilizând ca lichide de etalonare benzen şi acetonã.
În tabelul 5.1. se prezintã valorile experimentale ale constantelor electrice pentru
componenţii puri în intervalul de temperaturã 20-35 comparativ cu datele din literaturã.
Tabelul 5.1. Constantele dielectrice de 1-propanol şi MTBE
V.2. Metoda
experimentalã de determinare a
constantei dielectrice
Descrierea
instrumentului folosit la
mãsurare .
Pentru mãsurãtori se foloseşte dielometrul universal Hp OH 301, reprezentat în figura
5.1.
Procedura de efectuare a mãsurãtorilor este urmãtoarea:
1. Punerea la pãmânt a instrumentului:
Înainte de conectarea instrumentului la reţea trebuie controlat daca comutatorul de
tensiune este pe poziţia corectã.
Se verificã poziţia zero a aparatului . Dacã este nevoie se va ajusta poziţia mecanicã
de zero cu ajutorul şurubului de plastic din partea frontalã a instrumentului.
Se conecteazã instrumentul la reţea.
Se aduc scalele '' c'' şi ''G'' la zero. Comutatorul Range pe poziţia ''c'' şi comutatorul
senzitivity în poziţia 1.
Pagina 72 din 82
Substanţa Temperatura
20 25 30 35
1-propanol
MTBE

Se deschide instrumentul şi becul de semnalizare roşu se aprinde . Dupã douã
minute se aduce acul de la butonul ''Oc'' la zero roşu.
Dupã alte 15 minute instrumentul s-a încalzit
Indicatorul se aduce din nou cu ''Oc'' pe zero roşu.
Comutatorul se aduce în poziţia ''G'' , şi acul indicator este ajustat de la butonul
''O,G'' la zero roşu.
Comutatorul Range se aduce în poziţia ''10G'' şi cu butonul '' '' se aduce la zero
roşu.
Comutatorul Range se readuce în poziţia zero .
Instrumentul este gata de funcţionare.
2. Determinarea constantei dielectrice.
Mãsurãtoarea constantei dielectrice se realizezã prin determinarea variaţiei capacitãţii
unui condensator cu aer, dacã aerul utilizat ca dielectric este înlocuit cu substanţa de cercetat.
Figura 5.1 Dielcometrul universal OH 301
1. Scalã de la 0-110 pF a condensatorului variabil ''c'' .
2. Butonul condensatorului ''c''.
3. Comutatorul domeniului de lucru ''RANGE''.
4. Butonul de compensare ''Go''.
5. Bec de semnalizare .
6. Buton de compensare ''1Go''.
7. Comutator de reţea.
8. Buton de compensare ''10Go'
9. Comutator de sensibilitate '' sensitivity''.
10. Butonul condensatorului variabil G.
11. Scala 0-55 a condensatorului G.
Pagina 73 din 82

12. Instrument de nul.
13. Conexiuni la reţea
X - şuruburi.
V.3. Constantele dielectrice ale soluţiilor de 1-propanol şi MTBE
În tabelul 5.2 se prezintã rezultatele experimentale mãsurãtorilor de constante
dielectrice ale soluţiilor de 1-propanol şi MTBE la temperaturile 20, 25, 30, şi 35 .
Tabelul 5.2. Constantele dielectrice ale substanţelor de 1-propanol (x) şi MTBE în
intervalul 20-35 .
x
Temperaturã
20 25 30 35
0.0000 6.79 6.51 6.27 5.96
0.1517 7.34 6.92 6.73 6.58
0.3040 8.16 7.75 7.51 7.35
0.4094 9.10 8.90 8.60 8.44
0.5002 10.36 9.98 9.56 9.06
0.6095 11.62 11.33 10.92 10.61
0.7575 16.10 14.71 14.23 13.73
0.9099 18.63 18.60 16.96 15.88
1.0000 20.56 20.01 17.56 16.83
Variaţia constantei dielectrice în funcţei de concentraţia soluţiei la temperaturile de
20, 25, 30, şi 35 este ilustratã în figura 5.1.
Se pot face urmãtoarele observaţii:
Constanta dielectricã a soluţiilor studiate scade cu creşterea temperaturii.
La temperaturi constante constanta dielectricã creşte odatã cu creşterea
concentraţiei de 1-propanol, creşterea fiind cu atât mai accentuatã cu cât soluţiile sunt mai
concentrate în alcool.
Pagina 74 din 82

Figura 5.1 Variaţia constantei dielectrice cu concentraţia şi temperatura pt soluţiile de 1-
propanol (x) şi MTBE.
V.4. Polarizarea molecularã a soluţiilor de 1-propanol şi MTBE
Determinãrile experimentale de constantã dielectricã şi de densitate au permis calculul
polarizãrii moleculare a soluţiilor cu relaţia Clausius-Mossotti:
unde:
- polarizarea molecularã
- constanta dielectricã.
Pagina 75 din 82

M - masa molecularã medie a soluţiilor.
d - densitatea soluţiei.
În figura 5.2 se prezintã variaţia polarizãrii moleculare a soluţiilor de 1-propanol şi
MTBE în funcţie de concentraţia soluţiilor la temperaturile 20, 25, 30, şi 35 . Se observã
scãderea polarizãrii moleculare odatã cu creşterea concentraţiei de 1-propanol ceea ce este în
acord cu polarizabilitatea mai micã a alcoolului.
S-au calculat de asemenea valorile polarizãrii moleculare de exces
unde:
- polarizarea molecularã a soluţiilor şi .
şi - fracţiile molare ale componenţiilor 1şi 2.
- polarizarea molecularã a componenţilor 1 şi 2
Tabelul 5.3.1 Polarizarea molecularã a soluţiilor de 1-propanol şi MTBE
x PM 20 25 30 35
0,0000 78,3281 77,3790 76,6240 75,6996
0,1517 75,8383 74,3745 74,3009 74,2215
0,3040 73,9479 73,1990 73,1652 72,9015
0,4094 72,9943 72,9778 72,6308 72,3649
0,5002 72,7239 72,4831 72,0421 71,3626
0,6095 71,2425 71,1728 70,9578 70,8453
0,7575 70,9088 70,1696 70,0936 70,0254
0,9099 67,2240 67,0108 66,9175 66,6526
1,0000 64,8190 64,2421 64,0061 63,9559
Tabelul 5.3.2 Polarizare moleculara de exces a solutiilor de 1-propanol şi MTBE
x PME 20 25 30 35
0,0000 0 0 0 0
0,1517 8,9700 8,1095 8,3804 8,4884
0,3040 5,0221 4,9633 5,3230 5,3755
Pagina 76 din 82
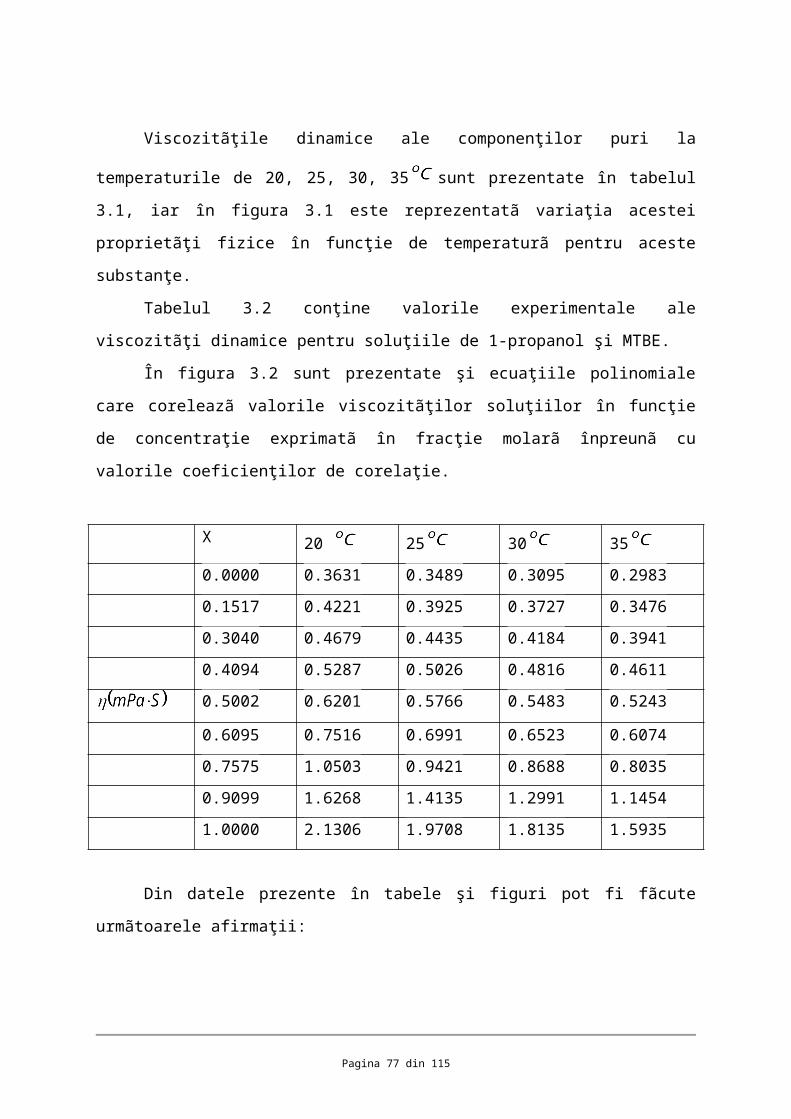
0,4094 2,6447 3,3575 3,4587 3,5981
0,5002 1,1476 1,6699 -1,7244 1,5325
0,6095 -1,8393 -1,1063 -0,7390 -0,8749
0,7575 -4,1433 -4,0237 -3,4706 -2,8264
0,9099 -9,8870 -9,1846 -8,5696 -7,9883
1,0000 0 0 0 0
În figura 5.3 se prezintã variaţia polarizãrii moleculare de exces în funcţie de
concentraţie de temperaturã .
Se pot evidenţia urmãtoarele aspecte:
Cele 4 curbe au forme similare de ''S''.
La concentraţii mici de alcool polarizarea molecularã de exces are valori
pozitive cu un maxim situat la +9,5.
La concentraţii mai mari de alcool , polarizarea molecularã de exces are
valori negative cu un minim situat la -9,5.
Pagina 77 din 82

Figura 5.1.Variaţia polarizării moleculare în funcţie de fracţia molară
a 1-propanolului (x) în intervalul de temperatură 20-35 °C.
Pagina 78 din 82

Figura 5.2.Variaţia polarizării moleculare de exces în funcţie de fracţia molară
a 1-propanolului (x) în intervalul de temperatură 20-35 °C.
Capitolul VI
Pagina 79 din 82
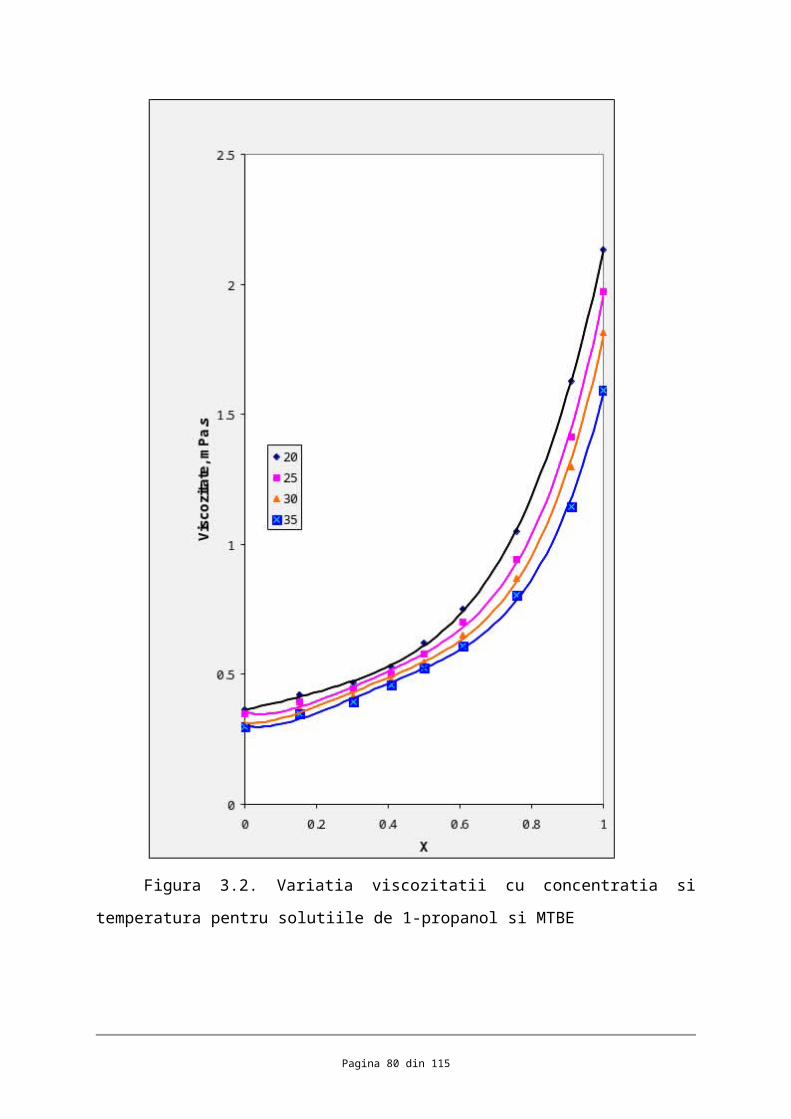
Concluzii
VI.1. Au fost determinate experimental viscozitãţile substanţelor pure şi ale soluţiilor
de 1-propanol şi MTBE cu fracţiile molare de alcool: 0.1517; 0.3040; 0.4094; 0.5002;
0.6095; 0.7575 şi 0.9099 în domeniul de tamperaturã 20-35 . S-a reprezentat grafic
variaţia viscozitãţii soluţiilor cu concetraţia şi temperatura evidenţiindu-se cã la concentraţie
constantã viscozitãţile soluţiilor scad odatã cu creşterea temperaturii, iar la temperaturã
constantã viscozitãţile acestor soluţii cresc odatã cu creşterea concentraţiei de alcool.
VI.2. S-au calculat valorile viscozitãţii de exces care au fost reprezentate grafic în
funcţie de temperaturã şi de concentraţia alcoolului şi au fost corelate cu ecuaţii polinomiale
de tip Redlich-Kister.
VI.3. Valorile viscozitãţii de exces sunt negative pe întreg domeniul de concentraţie şi
de temperaturã utilizat pentru toate sistemele studiate.
VI.4. Datele experimentale de viscozitate au fost reprezentate cu ajutorul urmãtoarelor
ecuaţii:
Dolezalek:
Grunberg-Nissan:
Wijk:
S-au trasat diagramele de variaţie a viscozitãţii calculate cu ecuaţiile de mai sus în
funcţie de concentraţie şi de temperaturã comparativ cu valorile mãsurate experimental
pentru toate soluţiile studiate în aceastã lucrare.
Pentru corelarea valorilor experimentale ale viescozitãţii soluţiilor studiate rezultã cã
ecuatia Wijk şi Dolezalek conduc la rezultatele cele mai bune.
VI.5. Coeficintul d din ecuaţia Grunberg-Nissan , corelat cu energia de interacţiune,
are valori negative pentru soluţiile cercetate şi aratã cã forţele de dispersie sunt importante în
sistemele studiate.
VI.6. Au fost calculate mãrimile de activare specifice procesului de curgere: energia
de activare şi factorul preexponeţial.
Energia de activare a curgerii este pozitivã pentru ambii componeţi puri
remarcându-se o valoare mai mare în cazul 1-propanol.
Factorul preexponeţial este de asemenea mai mare în cazul 1-propanol.
Pagina 80 din 82

Au fost determinate experimental constantele dielectrice ale componenţilor puri şi
ale soluţiilor de 1-propanol şi MTBE cu concentraţiile prezentate mai sus în intervalul de
temperaturã 20-35 .
VI.7. S-a reprezentat variaţia constantei dielectrice a soluţiilor cu concentraţia şi
temperatura, evidenţiindu-se cã la concentraţie constantã, constanta dielectricã creşte odatã cu
creşterea concentraţiei de alcool în acord cu polarizabilitatea mai mare a alcoolului
VI.8. S-au calculat valorile polarizãrii moleculare ale soluţiilor studiate precum şi
valorile polarizãrii moleculare de exces. Diagramele de variaţie a polarizãrii moleculare de
exces în funcţie de concentraţie şi temperaturã indicã valori pozitive ale acestei mãrimi de
exces la concentraţii mici de alcool, în timp ce la concentraţii mari de 1-propanol polarizarea
molecularã de exces are valori negative.
Capitolul VII
Aspecte metodologice
Sporirea eficienţei procesului de învãtãmant constituie o preocupare majorã.
Realizabilã printr-o multitudine de cãi şi mijloace, vizeazã printre altele ridicarea nivelului de
pregãtire atât a profesorului cât şi a elevului.
Ridicarea competenţei profesionale a cadrelor didactice, cât şi asigurarea unor indici
superiori de pregãtire a elevilor potrivit cerinţelor ştiinţei şi tehnicii contemporane constituie
premise ale unei eficienţe sporite.
Optimizarea procesului de învãţãmânt prin selectarea adevãrurilor ştiinţei dupã criterii
ca: esenţialitatea , caracterul formativ şi prospectiv, precum şi punerea de acord a logicii
didactice cu logica ştiinţificã trebuie sã fie asociate preocupãrii de accesibilitate a
cunoştinţelor transmise.
Prin accesibilitatea cunoştintelor înţelegem adecvarea calitativã şi cantitativã la
capacitãţile intelectuale ale elevilor. Accesibilitatea trebuie sã aibã în vedere atât
raţionalizarea volumului informaţional, cât şi asigurarea condiţiilor necesare pentru
recepţionarea lor. Ea este sprijinitã de intervenţii asupra conţinutului ca reconsiderarea
conţinuturilor tradiţionale (resistematizarea) şi asimilarea noilor achiziţii.
Pagina 81 din 82

Etapele respective sunt, în fapt, concomitente , ele interfereazã. La toate acestea se
adaugã modernizarea transmiterii, metode şi tehnici moderne de predare, activizarea elevilor ,
stimularea iniţiativei personale, gândirii creatoare.
Conducãtorul unei clase, profesorul, este în permanentã frãmântat de preocuparea de a
gãsi cel mai potrivit mod de a angaja elevul intr-o serie de activitãţi în mãsurã sã influenţeze
în cel mai înalt grad munca şi caracterul acestuia.
Este foarte dificil sã prezinţi ca un tot unitar toate aspectele parţiale, momentane pe
care le realizezi în organizarea şi conducerea unui cerc de chimie. Dincolo de întrebuinţãrile
şi recomandãrile ce ţi s-ar oferi, munca aceasta pãstreazã pregnant nota personalã a
profesorului, expresie a pregãtirii sale , a afinitãţii şi înclinaţiilor sale, a experienţei
dobândite.
Pagina 82 din 82